



Upper respiratory infections (URI) (i.e. those affecting the respiratory tract above the level of the vocal cords) are most commonly of viral aetiology, although bacteria may also be responsible( Reference Dasaraju and Liu 1 ). URI commonly result in work absence and primary care consultations and impose a major economic burden on society( Reference Fendrick, Monto and Nightengale 2 ). They also cause a majority of acute exacerbations of asthma and chronic obstructive pulmonary disease (COPD)( Reference Johnston 3 ), which represent the main cause of morbidity and mortality in people with these conditions. Moreover, they are frequently inappropriately treated with antibiotics( Reference Gulliford, Dregan and Moore 4 ), a practice which contributes to the emergence of antimicrobial resistance. In the absence of effective vaccines against the viruses that most commonly cause URI, identification of alternative preventive strategies is a research priority.
A growing body of evidence suggests that vitamin D supplementation may reduce the risk of URI in some clinical contexts. Vitamin D metabolites favourably modulate innate immune responses to respiratory viruses in vitro ( Reference Brockman-Schneider, Pickles and Gern 5 – Reference Telcian, Zdrenghea and Edwards 9 ) and numerous observational studies have reported independent inverse associations between circulating concentrations of 25-hydroxyvitamin D (25(OH)D, the major circulating vitamin D metabolite) and URI risk( Reference Jolliffe, Griffiths and Martineau 10 ). Meta-analyses of randomised controlled trials (RCT) of vitamin D supplementation for the prevention of acute respiratory infections and asthma exacerbations (which are commonly precipitated by URI) have reported protective effects that are strongest in those with the lowest circulating 25(OH)D concentrations at baseline( Reference Martineau, Cates and Urashima 11 – Reference Jolliffe, Greenberg and Hooper 13 ). However, scepticism regarding protective effects of vitamin D against URI remains: this may in part reflect limitations inherent in the design of cross-sectional and case–control studies, where the possibility of reverse causality cannot be excluded( Reference Jolliffe, Griffiths and Martineau 10 ). Genetic studies represent an alternative approach to assessing whether the vitamin D pathway plays a causal role in protection against URI; a key strength is that associations seen cannot be attributed to reverse causality. In terms of mechanism, SNP in the gene encoding the vitamin D receptor (VDR) may modify the protective efficacy of vitamin D-mediated host responses, for example, by influencing the structure of the VDR with consequences for transcription of vitamin D-regulated genes influencing immune function( Reference van Etten, Verlinden and Giulietti 14 ). VDR polymorphisms have previously been reported to associate with susceptibility to acute lower respiratory infections (LRI) in diverse settings( Reference Janssen, Bont and Siezen 15 – Reference Han, Hodemaekers and Nagarajah 18 ); to date, however, the influence of SNP in VDR on susceptibility to URI has not been investigated. Moreover, no study has investigated whether polymorphisms in other genes in the vitamin D pathway that encode proteins are responsible for vitamin D metabolism, transport and signalling associated with URI risk. We, therefore, conducted an investigation to test the hypothesis that one or more of the thirty-three SNP in eleven vitamin D pathway genes (Fig. 1) influence the risk of URI and are associated with pathogen-stimulated concentrations of cytokines and chemokines. The association between genotype and risk of URI was investigated in a discovery cohort of 725 adults, with validation of positive hits in a cohort of 737 children. Where genetic associations were replicated in both cohorts, ex vivo host responses to viral stimuli were compared between genetically susceptible v. resistant individuals.
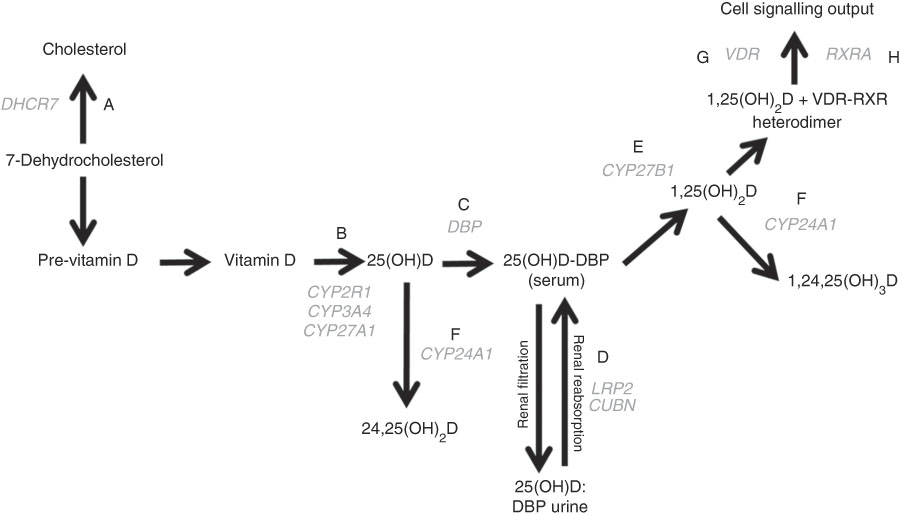
Fig. 1 The vitamin D pathway. DHCR7 (A) encodes the 7-dehydrocholesterol reductase enzyme, which catalyses the conversion of 7-dehydrocholesterol to cholesterol; CYP2R1, CYP3A4, and CYP27A1 (B) encode 25-hydroxylating cytochrome P450 enzymes; the vitamin D binding protein gene (DBP (C)) encodes the principal vitamin D transport protein; LRP2 and CUBN (D) encode the proteins megalin and cubilin, respectively, involved in renal re-absorption of 25-hydroxyvitamin D (25(OH)D) via receptor-mediated endocytosis; CYP27B1 (E) encodes the cytochrome P450 enzyme which 1-α-hydroxylates 25(OH)D to form 1,25-dihydroxyvitamin D (1,25(OH)2D); CYP24A1 (F) encodes the cytochrome P450 enzyme responsible for 24-hydroxylating vitamin D metabolites including 25(OH)D and 1,25(OH)2D; VDR (G) encodes the vitamin D receptor, which is ligated by 1,25(OH)2D and forms a heterodimer with the gene product of RXRA (H) – the retinoid X receptor – to mediate the biological actions of vitamin D. Gene symbols are shown in grey.
Methods
Participants
The discovery cohort comprised a total of 725 adults who participated in one of the three RCT of vitamin D supplementation to prevent acute respiratory infections conducted in London, UK, whose methods are described elsewhere( Reference Martineau, MacLaughlin and Hooper 19 – Reference Martineau, Hanifa and Witt 21 ). All studies were approved by East London and The City Research Ethics Committee 1 (Refs. 09/H0703/67, 09/H0703/76 and 09/H0703/112), and written informed consent was obtained from all participants before enrolment. The validation cohort comprised 737 children who were enrolled in the Manchester Asthma and Allergy Study (MAAS), a population-based birth cohort study whose methods are described elsewhere( Reference Custovic, Simpson and Murray 22 ). Children were recruited prenatally, and were born between 20 April 1995 and 13 April 2000. The study was approved by the Research Ethics Committee Greater Manchester East, NRES Committee (Ref. no. 14/NW/1309) and parents gave written informed consent for their children to participate.
Procedures
Clinical data collection
At enrolment, participants in the discovery cohort completed questionnaires detailing lifestyle and demographic factors relating to risk of URI including smoking history, influenza vaccination history, socio-economic position, age, sex and racial/ethnic origin. A baseline blood sample was collected from all participants for DNA extraction and isolation of serum for determination of 25(OH)D concentration. For a representative sub-set of 185 participants, a sample of whole blood was also incubated with a panel of Toll-like receptor (TLR) ligands and respiratory viruses for 24 h, as described in the forthcoming paragraphs. Participants were invited back 2 weeks after their baseline visit to be randomised to receive intervention or control regimens as summarised in online Supplementary Table S1 over a 1-year period, during which they completed a daily diary of URI symptoms. Symptom scores were used to define incident URI, as previously described, and validated in a subset of participants in the Vitamin D supplementation in prevention of influenza (ViDiFlu) trial( Reference Martineau, Hanifa and Witt 21 ) using PCR detection of eleven respiratory viruses obtained by nasopharyngeal swabs.
MAAS participants attended follow-up visits at the ages of 1, 3, 5, 8 and 11 years. At each visit, a validated questionnaire was used to collect information on incidence of URI diagnosed by a primary care physician. We extracted all data from primary care medical records, including URI, emergency department admissions and hospitalisations and prescriptions of asthma medications and oral corticosteroids( Reference Belgrave, Simpson and Semic-Jusufagic 23 ). Self-reported episodes were validated against primary care records. Blood samples were collected at age 11 years for peripheral blood mononuclear cell (PBMC) isolation and cryopreservation pending ex vivo stimulation( Reference Semic-Jusufagic, Belgrave and Pickles 24 ).
SNP selection in the discovery cohort
A literature search of the PubMed database was performed to identify SNP previously shown to associate with serum 25(OH)D concentration and/or risk of non-skeletal disease( Reference Jolliffe, Walton and Griffiths 25 ). A total of fifty-four such SNP in eleven genes in the vitamin D pathway were identified; their function is illustrated in Fig. 1. TagSNP were selected based on linkage disequilibrium information from the HapMap database (release #27: phases 1, 2 and 3 – merged genotypes and frequencies); ‘Utah residents with Northern and Western European ancestry from the Centre d'Etude du Polymorphisme Humain (CEPH) collection (CEU)’ dataset, represented the discovery cohort, of whom 83 % of participants were of White European ethnic/racial origin. Using Bioinformatics’ Haploview program (version 3.3), selecting the ‘pairwise tagging only’ option, setting the r 2 threshold to >0·8 and accepting a minimum genotype completeness of 75 % and a minor allele frequency threshold of 0·04, tagging reduced the number of alleles to be genotyped from fifty-four to thirty-seven SNP. Subsequent to closure of the HapMap database, tagging was re-checked using the 1000 genomes reference set under the same parameters, which highlighted two additional pairwise associations in the SNP panel. Thus, rs11568820 was chosen to capture rs7976091 (r 2 1·0), and rs1544410 was chosen to capture rs731236 (r 2 1·0), reducing the panel to thirty-five SNP.
DNA extraction and genotyping
For the discovery cohort, genotyping was conducted at the Genome Centre at Queen Mary University of London. DNA was extracted from whole blood using a salting-out method previously described( Reference Jolliffe, James and Hooper 26 ) on the Biomek FX robot (Beckman Coulter), quantified using the Nanodrop spectrophotometer (Thermo Scientific) and normalised to 5 ng/µl. In all, 10 ng DNA were used as template for 2 µl TaqMan assays (Applied Biosystems) performed on the ABI 7900HT platform in 384-well format and analysed with Autocaller software. Typing for two SNP failed (rs6127118, CYP24A1 and rs11574010, VDR), reducing the panel for analysis to thirty-three SNP.
For the validation cohort, genomic DNA was extracted from blood using the phenol–chloroform method and genotyped on an Illumina 610 quad array (Illumina) and quality controlled as described previously( Reference Paternoster, Standl and Waage 27 ). Genotypes were subsequently prephased (SHAPEIT version 2.5.1) and imputed (IMPUTE2 version 2.3.2) with the 1000 genomes haplotypes – phase 3 integrated variant set reference genotypes. In all, one SNP was genotyped (rs4334089) and three SNP were imputed (rs11568820, rs7970314 and rs2740574); their info scores( Reference Zheng, Rong and Liu 28 ) were 0·99, 0·99 and 0·97, respectively.
Whole blood assays
Stocks of rhinovirus (RV)-1B, RV-16 and respiratory syncytial virus (RSV) were prepared as previously described( Reference Telcian, Zdrenghea and Edwards 9 ). Samples of whole blood from a sub-set of participants in the discovery cohort were stimulated with the following TLR ligands and pathogens in sterile ninety-six-well polystyrene microplates (Corning Incorporated): polyinosinic:polycytidylic acid (poly I:C; InvivoGen; working stock concentration 1 mg/ml), resiquimod (R848; InvivoGen; working stock concentration 10 µg/ml), RV-1B (working stock titre 8·8×107 tissue culture infective dose 50 % (TCID50)/ml), RV-16 (working stock titre 9·8×107 TCID50/ml) and RSV (working stock titre 4×106 plaque-forming units/ml). In all, 180 μl of whole blood was incubated with 20 μl of a solution/suspension of the working stock of stimulant or vehicle (PBS) in a humidified incubator at 37°C and 5 % CO2 for 24 h. Supernatants were then aspirated and stored at −80°C pending analysis by multiplex ELISA (Human Cytokine Magnetic 30-Plex Panel; Invitrogen), following manufacturer’s instructions. Concentrations of the following inflammatory mediators were determined: IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8 (C-X-C motif ligand (CXCL)-8), IL-10, IL-12, IL-13, IL-15, IL-17, IL-1RA, IL-2R, interferon (IFN)-α, IFN-γ, TNF, C-C motif ligand (CCL)-2 (monocyte chemoattractant protein-1), CCL3 (macrophage inflammatory protein (MIP)-1α), CCL4 (MIP-1β), CCL5 (RANTES), CCL11 (eotaxin-1), CXCL9 (monokine induced by IFN-γ; MIG), CXCL10 (inducible protein (IP)-10), epidermal growth factor (EGF), fibroblast growth factor (FGF)-basic, hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF).
Peripheral blood mononuclear cell assays
Methods of the PBMC assays performed in the validation cohort are described in detail elsewhere( Reference Semic-Jusufagic, Belgrave and Pickles 24 ). Briefly, cryopreserved PBMC were thawed and distributed in ninety-six-well plates (2×105 cells per well) before stimulation with respiratory viruses (RV1B, RV16 and RSV, all at a multiplicity of infection (ratio of infectious agents to infection targets) of 1), resiquimod (1 µm final concentration) or medium. Supernatants were harvested 24 h after stimulation and concentrations of IFN-α, IFN-γ, CXCL10, IL-1β, IL-6 and TNF were measured with the Meso Scale Discovery Multi-array.
Measurement of vitamin D status
Serum concentrations of 25(OH)D2 and 25(OH)D3 were determined by isotope-dilution liquid chromatography–tandem MS in the Department of Clinical Biochemistry at Homerton Hospital, and summed to give total serum 25(OH)D concentration. The limit of detection for 25(OH)D2 and 25(OH)D3 was 10 nmol/l; mean percentage bias was −0·70 (sd 7·35); 95 % limits of agreement were from −15·1 to 13·7. Undetectable results were treated as zero values. This laboratory participates in the international vitamin D external quality assurance scheme (www.deqas.org/).
Statistical analyses
Statistical analyses were conducted using STATA version 12 and SNPTEST version 2.5.1. The influence of genotype on rate of URI or proportion with one or more URI was analysed with negative binomial regression and the Cochran–Armitage test, respectively. All SNP were analysed using the per-allele method, that is, under an additive model. Regression models for the discovery cohort were adjusted for the following potential confounders of the relationship between genotype and URI risk: age, sex, racial/ethnic origin, influenza vaccination history, smoking history, baseline vitamin D status, allocation to vitamin D v. placebo and presence of respiratory morbidity (asthma v. COPD v. none). Covariates were classified as categorical variables, with the exception of age and baseline vitamin D status, which were fitted as continuous variables. In the validation cohort, participants whose parents identified their ethnic origin as being other than ‘White European’ (3 % of the study population) were excluded from genetic analyses. Ethnicity was controlled for in the statistical analysis of the discovery cohort and a sensitivity analysis excluding data from participants who identified their ethnic origin as being other than ‘White European’ (15 % of the study population) was performed for significant SNP–URI risk associations. An additional sensitivity analysis removing participants in the intervention arm was performed on significant SNP–URI risk associations in the discovery cohort to account for variation in serum response to vitamin D supplementation. Supernatant concentrations of inflammatory mediators were transformed to their natural logarithms before regression analysis. Regression coefficients were then exponentiated to give adjusted geometric mean ratios (aGMR) with associated 95 % CI and P values for trend. The Benjamini–Hochberg procedure for multiple testing correction was applied to analyse URI rate and immunological data to control the false discovery rate (FDR) at 10 %.
Results
Study populations
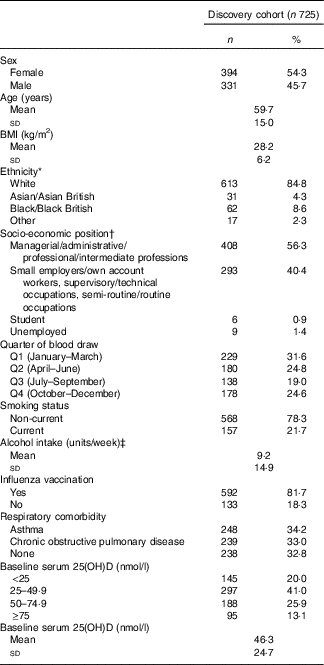
Characteristics of the 725 participants in the discovery cohort are presented in Table 1. They ranged from 16 to 94 years of age, with a mean of 59·7 (sd 15·0) years, and 54 % were female. In all, 67 % of participants had respiratory comorbidity (34 % asthma; 33 % COPD). The majority of participants classified their racial/ethnic origin as ‘White European’ (85 %); 9 % classified their racial/ethnic origin as Black/Black British; 4 % as Asian/Asian British and 2 % as mixed. At baseline, the mean serum 25(OH)D concentration was 46·3 (sd 24·7) nmol/l. Average duration of participation was 0·93 years, and participants had a total of 677 participant-years of follow-up. Immunological data were available for 180 participants in the discovery cohort; characteristics of this sub-set of participants were similar to those of participants who did not contribute immunological data (online Supplementary Table S2). Detailed characteristics of the replication population are presented elsewhere( Reference Semic-Jusufagic, Belgrave and Pickles 24 ) and summarised in online Supplementary Table S3. Briefly, 737 participants had data on URI and genotyping data. Average duration of participation was 9·97 years, and participants had a total of 7350 participant-years of follow-up. PBMC stimulation data and genotyping data were available for 228 children.
Table 1 Baseline participant characteristics, discovery cohort (Numbers and percentages; mean values and standard deviations)
25(OH)D, 25-hydroxyvitamin D.
* Ethnicity not reported in n 3. Other ethnicities: n 7 White and Black Caribbean, n 2 White and Asian, n 2 White and Black African, n 1 Irish – Sri Lankan, n 1 Spanish – Filipino, n 1 Asian Caribbean, n 1 Mauritian, n 2 preferred not to disclose.
† Socio-economic position not reported in n 9.
‡ Alcohol consumption not reported in n 13. One alcohol unit=8 g pure alcohol.
Genetic determinants of upper respiratory infections risk
Results of statistical analyses to determine the influence of genotype on risk of URI in the discovery cohort are presented in Table 2. After adjusting for potential confounders and correcting for multiple comparisons testing, carriage of minor alleles for three SNP in VDR and one SNP in CYP3A4 were found to associate with increased URI risk. Adjusted incidence rate ratios (aIRR) per additional minor allele, generated by additive models, are as follows: for rs4334089 in VDR, aIRR=1·15, 95 % CI 1·01, 1·31, P=0·03; for rs11568820 in VDR, aIRR=1·23, 95 % CI 1·07, 1·40, P=0·002; for rs7970314, aIRR=1·17, 95 % CI 1·02, 1·34, P=0·018; and for rs2740574 in CYP3A4, aIRR=1·27, 95 % CI 1·03, 1·56, P=0·025. Results of two sensitivity analyses to determine the influence of rs4334089, rs11568820, rs7970314 and rs2740574 on risk of URI, restricted to participants of white European ethnicity and the placebo arm, are presented in online Supplementary Tables S4 and S5, respectively. All SNP–URI associations remained significant (P≤0·048) for both sensitivity analyses. We also performed an exploratory interaction analysis to determine whether the influence of rs4334089 genotype on risk of URI in discovery cohort participants was modified by baseline vitamin D status (25(OH)D <25 v. ≥25 nmol/l). We found no evidence of effect modification (P value for interaction=0·38).
Table 2 Genetic determinants of upper respiratory infection risk, discovery cohort (Numbers and 95 % confidence intervals)
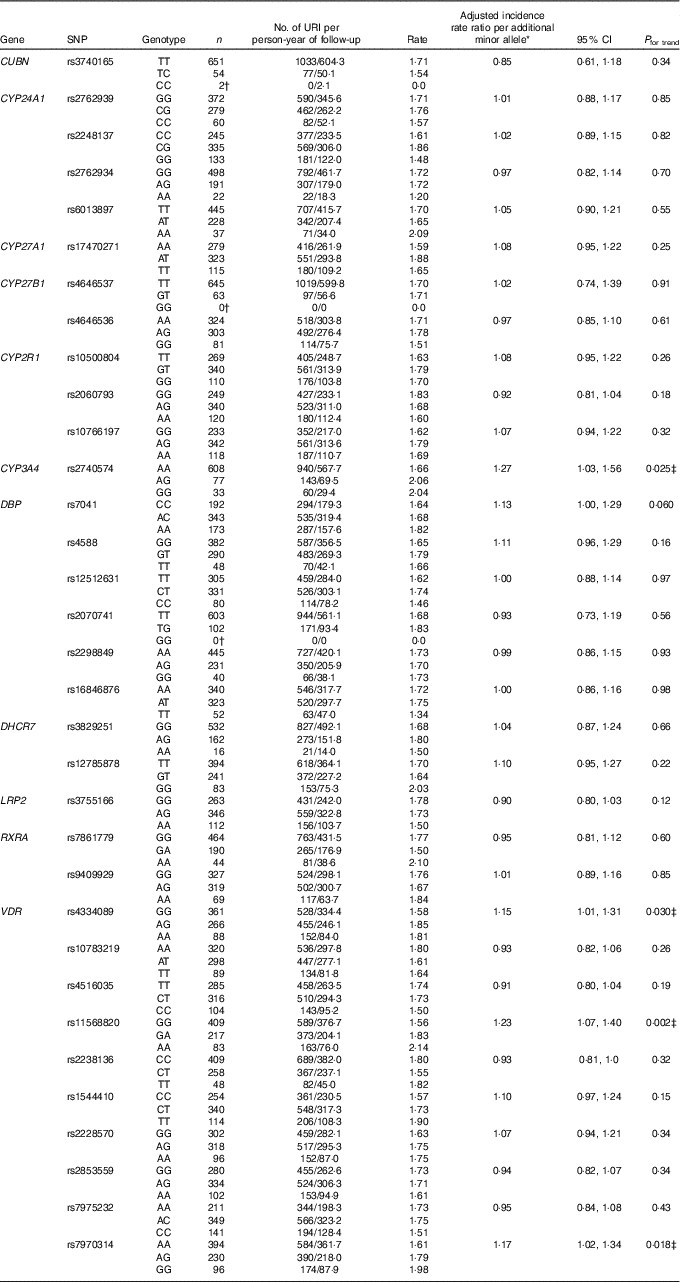
URI, upper respiratory infection; CUBN, cubilin; CYP-, cytochrome P450 enzyme; DBP, vitamin D binding protein; DHCR7, 7-dehydrocholesterol reductase enzyme; LRP2, low density lipoprotein-related protein 2 (also known as megalin); RXRA, retinoid-X receptor-A; VDR, vitamin D receptor; COPD, chronic obstructive pulmonary disease.
* Adjusted for age, sex, ethnicity, smoking history, influenza vaccination history, allocation to vitamin D v. placebo, respiratory comorbidity (asthma v. COPD v. none).
† URI rate could not be calculated due to zero participants with the genotype or zero URI events.
‡ Significant after correction for multiple comparisons testing, using the Benjamini and Hochberg method with a false discovery rate of 10 %.
We then proceeded to investigate whether these SNP also associated with susceptibility to URI in the validation cohort of children. Results of these analyses are presented in Table 3. The association between carriage of the minor allele for rs4334089 and increased susceptibility to URI originally observed in the discovery cohort was replicated in the validation cohort: the proportion of children experiencing at least one physician-diagnosed URI with AA v. AG v. GG genotypes was 6/56 (10·7 %) v. 23/278 (8·3 %) v. 21/403 (5·2 %), respectively (P for trend=0·048). No associations between imputed genotypes for rs11568820, rs7970314 or rs2740574 and URI susceptibility were seen in the validation cohort.
Table 3 Genetic determinants of upper respiratory infection risk, validation cohort
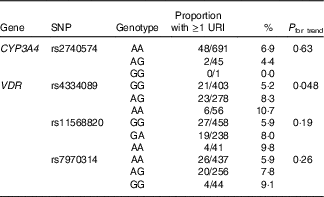
URI, upper respiratory infection; CYP-, cytochrome p450 enzyme; VDR, vitamin D receptor.
Genetic variation in ex vivo virus-stimulated inflammatory profiles
Having replicated the association between rs4334089 genotype and susceptibility to URI in separate cohorts of children and adults, we proceeded to investigate whether genetic variation at this locus was also associated with differences in host response to ex vivo viral stimuli in both discovery and validation cohorts. In the discovery cohort, concentrations of thirty inflammatory mediators in supernatants of virus-stimulated whole blood were compared between three groups of participants defined by their rs4334089 genotype (GG v. AG v. AA). After correction for potential confounders, statistically significant differences in supernatant concentrations of four inflammatory mediators were found (Fig. 2). Carriage of the minor allele of rs4334089 – previously shown to associate with increased susceptibility to URI – associated with lower supernatant concentrations of IL-2R (RSV stimulation: aGMR 0·92, 95 % CI 0·84, 0·99, P for trend=0·047; poly I:C stimulation: aGMR 0·89; 95 % CI 0·79, 0·99; P=0·041), CCL5 (RV1-B stimulation: aGMR 0·66, 95 % CI 0·46, 0·94, P for trend=0·023), CXCL9 (RV-16 stimulation: aGMR 0·84, 95 % CI 0·71, 1·00; P for trend=0·054) and IL-1RA (RSV stimulation: aGMR 0·91, 95 % CI 0·83, 0·99; P for trend=0·042). rs4334089 genotype also influenced host response in the MAAS validation cohort of children, where carriage of the minor allele was associated with lower supernatant concentration of resiquimod-stimulated CXCL10 (GMR 0·78, 95 % CI 0·64, 0·96; P for trend=0·017; Fig. 2). However, none of these differences remained statistically significant after correction for multiple comparisons testing, using the Benjamini and Hochberg method with a FDR of 10 %.
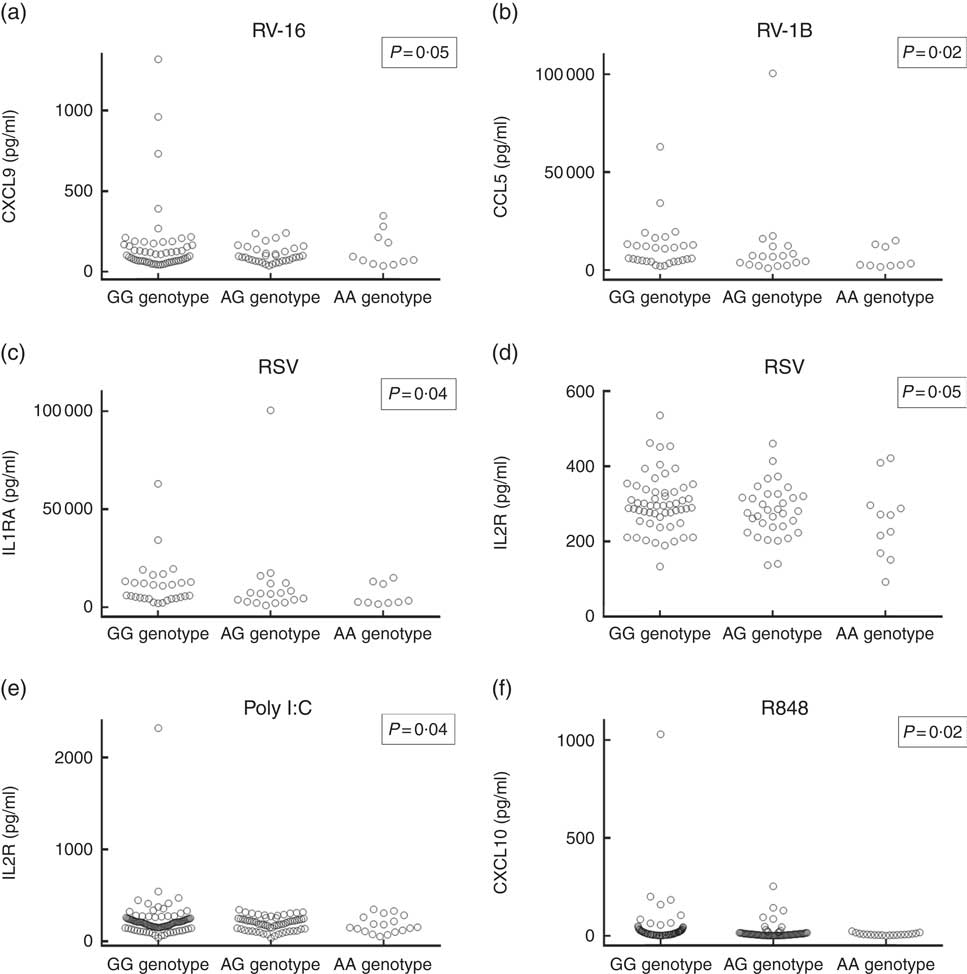
Fig. 2 Concentrations of virus-stimulated immune mediators by rs4334089 genotype. (a) Rhinovirus (RV)-16-stimulated C-X-C motif ligand (CXCL)-9, whole blood supernatant (discovery cohort, n 101); (b) RV-1B-stimulated C-C motif ligand (CCL)-5, whole blood supernatant (discovery cohort, n 56); (c) respiratory syncytial virus (RSV)-stimulated IL-1RA, whole blood supernatant (discovery cohort, n 101); (d) RSV-stimulated IL-2R, whole blood supernatant (discovery cohort, n 101); (e) polyinosinic:polycytidylic acid (poly I:C)-stimulated IL-2R, whole blood supernatant (discovery cohort, n 180); (f) resiquimod-stimulated CXCL10, peripheral blood mononuclear cell supernatant (validation cohort, n 228). P values are from regression analysis of log-transformed data; none was significant after correction for multiple comparisons testing, using the Benjamini and Hochberg method with a false discovery rate of 10 %.
Discussion
To our knowledge, this is the first study to investigate the influence of genetic variation in the vitamin D pathway on URI risk. We report that carriage of minor alleles for three SNP in VDR (rs4334089, rs11568820 and rs7970314) and one SNP in CYP3A4 (rs2740574) associated with increased risk of URI in a cohort of adults after correction for potential confounders and multiple comparison testing; these associations were robust to sensitivity analysis restricted to those who classified their ethnic origin as being ‘White European’. We achieved partial replication of these findings by additionally demonstrating an association between carriage of the minor allele for rs4334089 and increased susceptibility to URI in a validation cohort of children. Since the function of VDR is specific to vitamin D signalling, our results complement the growing body of evidence from in vitro studies, observational epidemiology and RCT suggesting that vitamin D plays a role in defence against viral respiratory infections.
Our study adds to the body of literature reporting associations between VDR polymorphisms and susceptibility to diverse infectious diseases, including LRI( Reference Janssen, Bont and Siezen 15 – Reference Han, Hodemaekers and Nagarajah 18 , Reference Jolliffe, Walton and Griffiths 29 ). The mechanisms underlying such associations have only been elucidated in some cases: for example, the mutant allele of the FokI polymorphism encodes a shorter VDR protein that results in higher NF-κB-driven transcription, enhanced expression of IL-12 and more vigorous lymphocyte proliferation in response to phytohaemagglutinin( Reference van Etten, Verlinden and Giulietti 14 ). In the absence of evidence that the rs4334089 polymorphism has similar functional consequences, we presume that the association demonstrated here arises as a result of this SNP being in linkage disequilibrium with a functional variant elsewhere in the VDR gene that has yet to be identified. Other studies have reported associations between the rs4334089 genotype and susceptibility to Parkinson’s and Alzheimer’s diseases( Reference Butler, Burt and Edwards 30 ), as well as responsiveness to vitamin D supplementation in patients with tuberculosis( Reference Ganmaa, Munkhzul and Fawzi 31 ).
Our immunological findings also have biological plausibility: since vitamin D modulates TLR expression via VDR signalling( Reference Sadeghi, Wessner and Laggner 32 – Reference Hansdottir, Monick and Lovan 34 ), it follows that genetic variation at the VDR has potential to influence TLR ligand-stimulated responses. With regard to specific cytokines and chemokines, IL-1RA plays a key role in resolution of RV infection( Reference Yoon, Zhu and Gwaltney 35 , Reference Hudock, Liu and Mei 36 ), while CCL5 has been reported to inhibit RSV infection in a human respiratory epithelial cell line( Reference Elliott, Tebbey and Pryharski 37 ) and to promote RSV clearance in mice( Reference Culley, Pennycook and Tregoning 38 ). CCL5, CXCL9 and CXCL10 recruit T lymphocytes in the context of respiratory virus infection( Reference Elliott, Tebbey and Pryharski 37 – Reference Lindell, Lane and Lukacs 40 ). Reduced production of these mediators in response to viral stimuli might, therefore, be expected to increased susceptibility to URI. However, the fact that genotypic differences in host response to viral stimuli did not remain statistically significant after correction for multiple comparisons testing indicates that these may have arisen due to type 1 error, and our immunological findings should not, therefore, be given undue weight.
Our study has several strengths. To our knowledge, it represents the most comprehensive investigation into the influence of genetic variation in the vitamin D pathway on susceptibility to any infectious disease that has been conducted to date, in terms of the number of vitamin D pathway genes investigated. Any attendant potential for type 1 error in genetic analyses was minimised by stringent adjustment for multiple comparisons. We handled the potential for confounding by adjusting for potential confounders, and by conducting appropriate sensitivity analyses. We utilised a PCR-validated definition of URI, and captured events prospectively using a detailed symptom diary over a 1-year period. Our study population incorporated a broad range of participants, with a wide range of circulating 25(OH)D concentrations, with and without respiratory comorbidity, which enhances generalisability of our findings. The fact that we were able to replicate the association between rs4334089 genotype and susceptibility to URI in diverse clinical cohorts (children v. adults), using a different case definition for URI (physician-diagnosed URI v. diary-defined URI), adds weight to this finding.
Our study also has some limitations. Our results are associative and descriptive; until a mechanism by which this VDR polymorphism influences susceptibility to URI is demonstrated, causal inferences should not be drawn. Our immunological studies investigated host responses in peripheral blood only; these may not reflect responses in the airway. Moreover, immunological data were only available for a sub-set of participants, and consequently the numbers of minor homozygotes contributing to analyses of inflammatory mediator concentrations were small: power to detect small differences in host response by genotype was, therefore, limited. It could be argued that our discovery and validation cohorts were too different in terms of age range to represent a true replication. However, the virology of URI in schoolchildren and adults in the UK is broadly comparable, in that RV are responsible for the majority of events in both populations( Reference Nicholson, Kent and Hammersley 41 , Reference Horn, Brain and Gregg 42 ). Moreover, vitamin D supports broad-spectrum innate antiviral responses, which are effective against different respiratory viruses( Reference Greiller and Martineau 7 ). Our finding that genetic variation in VDR associates with susceptibility to URI in both children and adults is also consistent with findings from our recent individual participant data meta-analysis of randomised trials, which showed an overall protective effect of vitamin D supplementation against URI in participants aged 0–95 years, with strongest effects seen in those with the lowest 25(OH)D levels receiving daily or weekly vitamin D supplementation( Reference Martineau, Jolliffe and Hooper 12 ). In the validation cohort, it is likely that only a sub-set of URI were captured, since only relatively severe episodes of URI are likely to present to primary care. However, the fact that captured episodes were physician-diagnosed provides extra confidence that diagnoses were accurate. Our study was also limited by the fact that we did not characterise viruses responsible for URI episodes; accordingly, we were unable to investigate whether VDR genotype influences susceptibility to URI in a pathogen-specific manner. Further research incorporating virological diagnosis could provide insight into this question; such studies may be challenging, as sensitivity of PCR for virus detection in URI can be <50 %, particularly when sampling is delayed after onset of symptoms( Reference Jiang, Lee and Cui 43 ). Finally, we draw attention to the fact that the vitamin D status of participants in our discovery cohort was relatively low (20 % <25 nmol/l, 61 % <50 nmol/l): if the genetic associations reported are modified by circulating concentrations of 25(OH)D, they may not be replicated in populations with higher vitamin D status.
In conclusion, we report that a polymorphism in VDR independently associates with susceptibility to URI in both adults and children. Given that the VDR is exclusively involved in vitamin D signalling, our findings lend weight to the growing body of evidence suggesting a role for the vitamin D pathway in mediating protection against respiratory infections.
Acknowledgements
The authors thank Dr Peter Timms, Ms Marion Rowe and Mr Tim Venton (Homerton Hospital, London) for performing 25(OH)D assays and Professor Seif Shaheen (Queen Mary University of London, UK) for his comments on this manuscript.
This is a summary of independent research funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Ref. no. RP-PG-0407-10398). The views expressed are those of the authors and not necessarily those of the National Health Service (NHS), the NIHR or the Department of Health. The work was also supported by a Chair from Asthma UK (no. CH11SJ) and Medical Research Council Centre (grant no. G1000758). S. L. J. and R. T. W. are NIHR senior investigators. MAAS was supported by the Asthma UK grants no. 301 (1995–1998), no. 362 (1998–2001), no. 01/012 (2001–2004), no. 04/014 (2004–2007), the BMA James Trust, The Moulton Charitable Foundation (2004–current); age 11 years clinical follow-up is funded by the Medical Research Council (MRC) UK (G0601361) and age 18 years follow-up by the Biotechnology and Biological Sciences Research Council/MRC (MR/L012693/1).
A. C. reports personal fees from Novartis, Regeneron/Sanofi, ALK, Bayer ThermoFisher, GlaxoSmithKline and Boehringer Ingelheim, outside the submitted work. N. C. B. is a shareholder and employee of GlaxoSmithKline. S. L. J. reports personal fees from Therapeutic frontiers, Myelo Therapeutics GmbH, Concert Pharmaceuticals, Bayer, Synairgen, Novartis, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Sanofi Pasteur, Centocor and Aviragen and holds patents for anti-virus therapies for respiratory diseases. A. R. M. reports grants from NIHR, during the conduct of the study. A. S. reports grants from medical research Council, JP Moulton Charitable Foundation and NIHR CRF, and personal fees from Thermo Fisher Scientific, during the conduct of the study.
A. R. M., D. A. J., C. J. G. and R. T. W. designed the discovery study. S. L. J., A. C., J. A. C. and A. S. designed the validation study. M. H., C. A. M., D. A. J. and J. A. C. developed and undertook genotyping assays. A. R. M., C. L. G. and D. A. J. developed and performed immunological assays in the discovery study. E. B. and A. G. T. developed and performed immunological assays in the validation study. D. A. J., J. A. C. and A. C. performed data analysis. A. R. M. and D. A. J. wrote the article; all other authors critically reviewed it and approved the final version.
The authors have no conflicts of interest to declare.
Supplementary material
For supplementary material/s referred to in this article, please visit https://doi.org/10.1017/S000711451800209X







