



Introduction
The DNA damage response (DDR) is essential to maintain genomic integrity in the face of a continuous onslaught of DNA damage from endogenous and environmental sources. Activation of this response involves the close coordination of DNA repair pathways and signalling to cell cycle arrest to allow repair and prevent DNA damage being copied (G1 and S-phase checkpoint) or transmitted to the next generation (G2/M checkpoint). The kinases ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia mutated and rad3 related (ATR) are DNA damage sensors that are at the apex of a phosphorylation and dephosphorylation cascade signalling to both cell cycle arrest via inactivation of cyclin-dependent kinases (CDKs) (Fig. 1), and DNA repair. ATM and ATR have overlapping but non-redundant activities with substantial cross-talk between the two pathways (Ref. Reference Weber and Ryan1). This review will describe the role of these signalling cascades and the development of drugs targeting them for anti-cancer therapy.
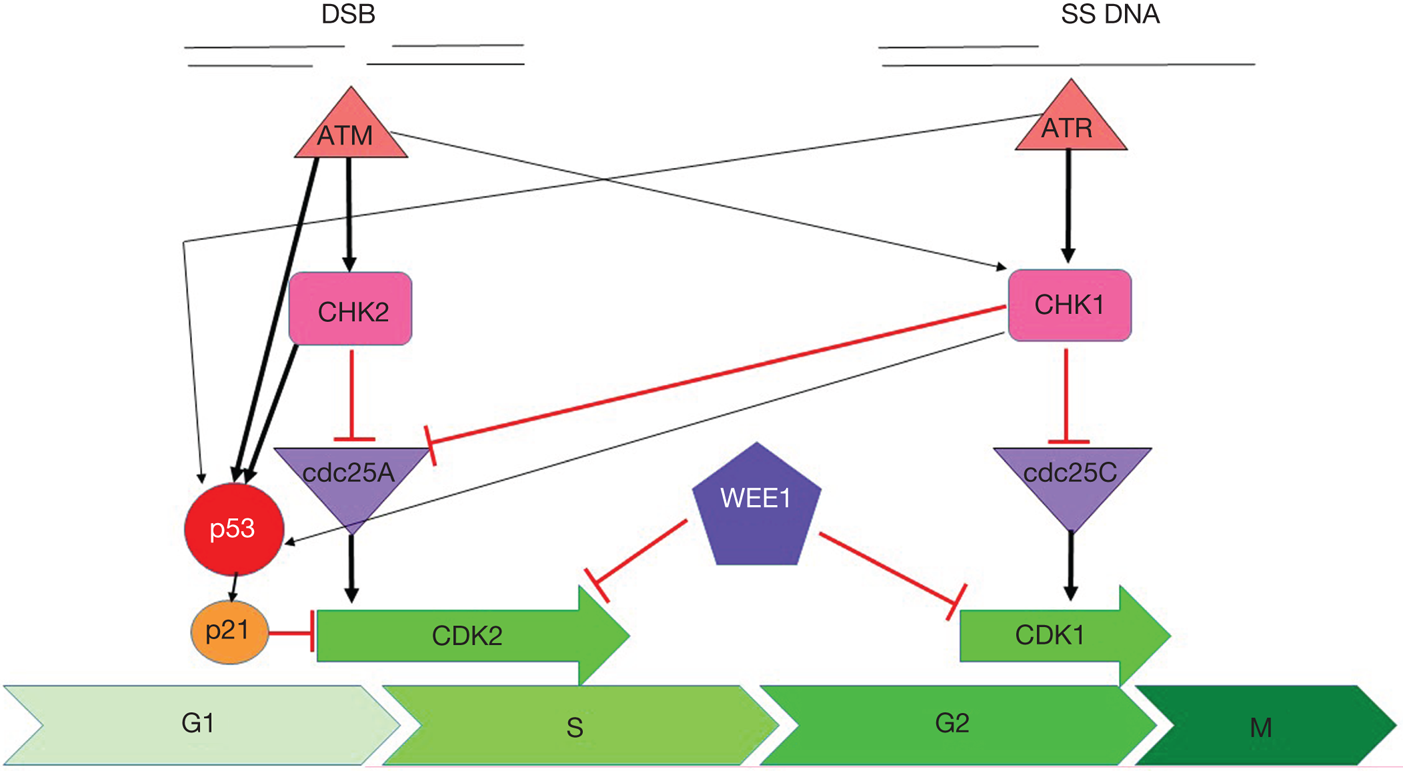
Fig. 1. Cell cycle checkpoint signalling. DNA double-strand breaks activate ATM, which phosphorylates and activates CHK2, which phosphorylates and inactivates cdc25A, preventing it from removing the inactivating phosphate on CDK2 thereby inhibiting S-phase entry and progression. Both ATM and CHK2 phosphorylate p53 resulting in transactivation of p21 to inhibit CDK2. SS-DNA (e.g. at stalled replication forks) activates ATR, which phosphorylates and activates CHK1, which phosphorylates and inactivates cdc25c, preventing it from removing the inactivating phosphate on CDK1 thereby inhibiting G2/M progression. There is substantial cross-talk between the two pathways with CHK1 also being a target of ATM and cdc25A a target of CHK1 and both ATR and CHK1 targeting p53. In addition, DNA damage activates WEE1 which phosphorylates and inactivates both CDK1 and CDK2. Black arrows indicate main activation pathways, grey ones are secondary pathways and red lines indicate inhibition.
The role of ATM/CHK2 pathway in cell cycle checkpoints
ATM is activated in response to DNA double-strand breaks (DSBs) (Ref. Reference Maréchal and Zou2). In undamaged cells, ATM exists as a dimer. Upon recruitment by the MRE11/RAD50/NBS1 (MRN) complex to DSBs, ATM autophosphorylates at serine 367 (ser367), serine 1893 (ser1893), serine 1981 (ser1981) and serine 2996 (ser2996) resulting in monomerisation and activation (Refs Reference Kozlov3, Reference Tanya4). Active ATM phosphorylates many target proteins regulating DNA repair, cell cycle arrest and apoptosis including CHK2, p53 and H2AX (Ref. Reference Shiloh and Ziv5). ATM plays a crucial role in the activation of the G1/S cell cycle checkpoint, primarily mediated through p53 activity. The most important transducer of ATM signalling is CHK2, a kinase that signals to DNA repair, cell cycle arrest and apoptosis. ATM phosphorylates CHK2 on threonine 68 (thr68) causing CHK2 dimerisation and autophosphorylation of the kinase domain, required for full activation (Ref. Reference Zannini, Delia and Buscemi6).
Active CHK2 phosphorylates the Cdc25A and Cdc25C phosphatases, which results in their inactivation/degradation. This promotes cell cycle arrest as active cdc25A/C remove inhibitory phosphorylation on CDKs that drive cell cycle progression. Cdc25A dephosphorylates CDK2, promoting progression into S phase. Cdc25C also dephosphorylates CDK1, which is usually held in the inactive state via phosphorylation by WEE1 and Myt1, promoting the transition into M phase (Refs Reference Weber and Ryan1, Reference O'Connell7, Reference Chow and Poon8). Although ATM can signal to G2 arrest via CHK2, the cell cycle defects observed in ATM-deficient cells are primarily G1/S checkpoint deficiency (Refs Reference Kastan9–Reference Weber11).
CHK2 also causes cell cycle arrest by phosphorylating the tumour suppressor p53 on ser15 and ser20 resulting in p53 stabilisation and activation (Ref. Reference Ou12). p53 is a transcription factor which, when active, initiates the transcription of genes involved in DNA repair, cell cycle arrest, apoptosis and metabolism as well as its own negative regulators (Ref. Reference Fischer13) for example, mouse double minute 2 (MDM2), a ubiquitin ligase that targets p53 for degradation (Ref. Reference Wade, Wang and Wahl14). In response to DNA damage, p53 is phosphorylated by many kinases including ATR and CHK1 as well as ATM and CHK2, contributing to cross-talk between the two pathways. This phosphorylation blocks the interaction between p53 and MDM2 leading to p53 protein accumulation (Ref. Reference Roos and Kaina15). Active p53 promotes the transcription of CDKN1A, which encodes the cyclin-dependent kinase inhibitor p21CIP1/WAF1 (Ref. Reference Abbas and Dutta16). p21 mediates p53-dependent G1 cell cycle arrest (Refs Reference Waldman, Kinzler and Vogelstein17, Reference Deng18). p53 activation also leads to the transcription of pro-apoptotic genes including Puma, Noxa, BAX and Apaf1, resulting in apoptotic cell death if the damage is sustained (Ref. Reference Bieging, Mello and Attardi19). The regulation of p53-mediated G1 checkpoint arrest and/or apoptosis by the DDR kinases is likely dependent on the context of the DNA damage, such as the type of DNA damage, cell cycle phase and molecular pathology of the cell type in question.
Figure 2 illustrates how the ATM/CHK2/p53 signalling pathway leads to cell cycle arrest and the maintenance of genome integrity. This pathway suppresses tumorigenesis and as a consequence, defects are often observed in cancer.
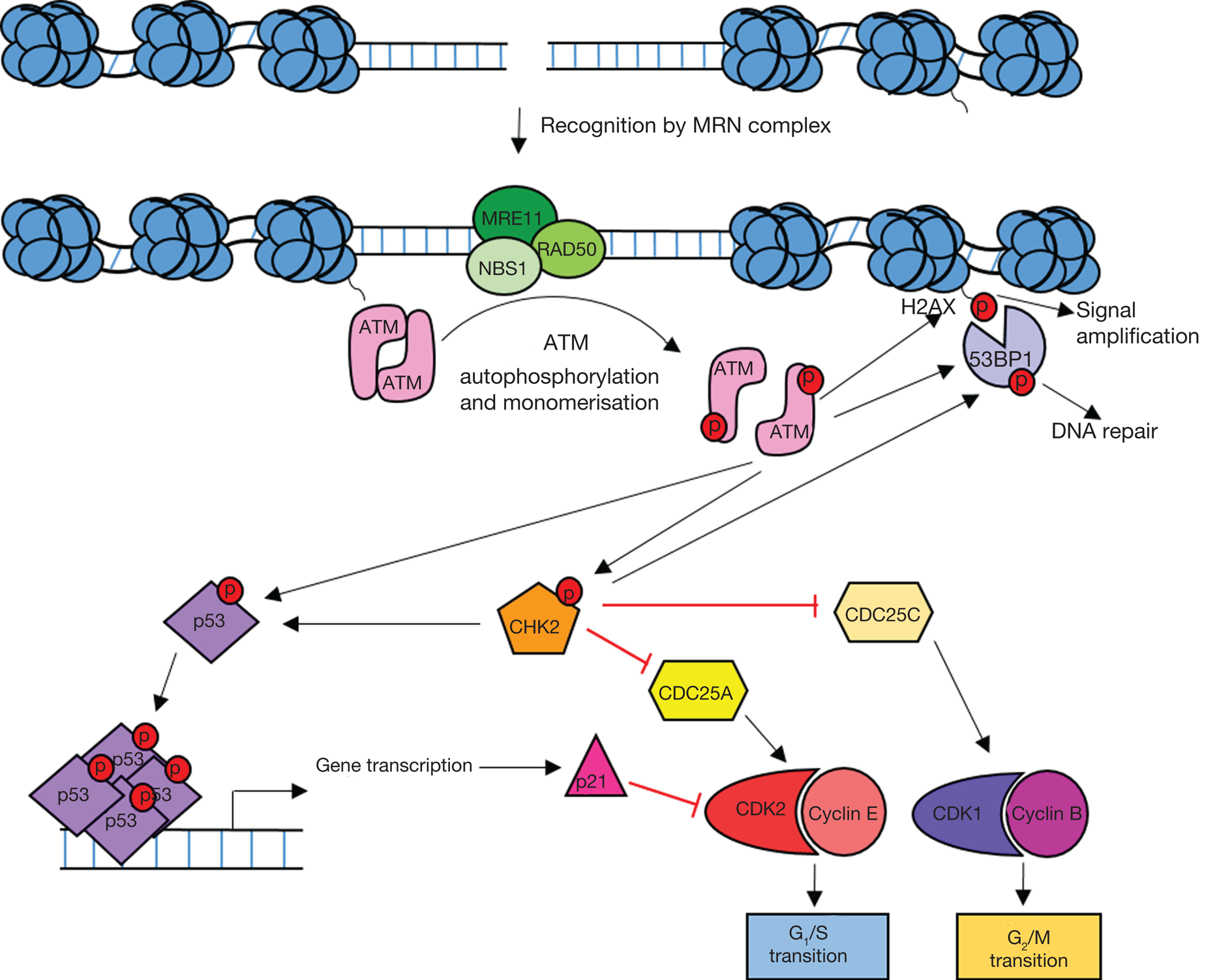
Fig. 2. Overview of ATM signalling in response to DNA damage. DNA double-strand breaks are recognised by the MRE11/RAD50/NBS1 (MRN) complex which recruits ATM leading to ATM activation. Active ATM phosphorylates the histone variant H2AX (γH2AX) leading to amplification and spreading of the damage signal. ATM-dependent phosphorylation of p53 and CHK2 leads to the activation of DNA repair processes and cell cycle arrest. Active p53 induces G1 arrest through transcriptional activation of the CDKN1A gene which codes for the cyclin-dependent kinase (CDK) inhibitor p21. Active CHK2 also phosphorylates p53 as well as CDC25 phosphatases resulting in S and G2 arrest. 53BP1 is recruited to yH2AX and phosphorylated by ATM and CHK2 leading to DNA repair.
Role of ATM/CHK2 in DNA repair
Although not essential for the repair of the majority of DSBs, ATM activity is required for the repair of a subset of DSBs generally associated with heterochromatin (Ref. Reference Goodarzi, Jeggo and Lobrich20). In response to DNA DSBs and stalled replication forks the variant histone H2AX is phosphorylated at ser139 by DNA-PK, ATM and ATR resulting in the accumulation of γH2AX in the vicinity of the DNA lesion. In ATM-deficient cells, around 10–15% of DNA damage foci, identified by antibodies against γH2AX, are retained 72 hours after ionising radiation (IR) (Ref. Reference Goodarzi21). γH2AX recruits mediator of DNA damage checkpoint 1 (MDC1) (Ref. Reference Scully and Xie22). Phosphorylation by ATM stabilises MDC1 on chromatin where it acts as a molecular scaffold recruiting chromatin modifiers to relax heterochromatin in the vicinity of the DSB (Ref. Reference Blackford and Jackson23). More MRN complexes are recruited to the site of the DSB by the interaction between MDC1 and NBS1, which in turn recruits and activates further ATM kinases, resulting in amplification of the signal along chromatin (Refs Reference Melander24, Reference Spycher25). ATM activation promotes DNA repair indirectly via both non-homologous end joining (NHEJ) and homologous recombination DNA repair (HRR), two prominent DSB repair pathways. ATM-dependent phosphorylation of 53BP1 recruited to damage markers on chromatin prevents DSB end resection, promoting NHEJ (Ref. Reference Panier and Boulton26). Conversely, ATM and CHK2 phosphorylate BRCA1 at the site of DNA damage (Ref. Reference Greenberg27). BRCA1 plays a critical role in the initiation of HRR (described in the section ‘Role in DNA repair’) (Ref. Reference Prakash28). 53BP1 and BRCA1 show mutual antagonism and, although both proteins are present throughout the cell cycle, pathway dominance is largely governed by the cell cycle stage and cyclin-CDK activity (Refs Reference Chen29, Reference Ceccaldi, Rondinelli and D'Andrea30).
In addition to BRCA1, ATM-mediated regulation of nucleases is required for efficient DNA end resection and activation of ATR at DSBs (Refs Reference Jazayeri31, Reference Myers and Cortez32). ATR signalling is discussed in the section ‘Role of ATR-CHK1-WEE1 in cell cycle checkpoints’. However, a direct role of ATM in DNA repair is unclear as ATM is dispensable for the repair of the majority of DNA DSBs, although it has an important role in initiating the chromatin remodelling cascade induced by phosphorylation of histone H2AX, and signalling to cell cycle checkpoint arrest (Ref. Reference Scully and Xie22).
Pathway dysfunction in cancer
Aberrations in the ATM gene are commonly seen in cancer. Homozygous germline mutations in ATM result in ataxia telangiectasia (A-T), a well characterised recessive genetic disease which predisposes to the development of cancer (Ref. Reference Rothblum-Oviatt33). Somatic mutations of ATM have been identified in many cancer types, most commonly lymphoid malignancies, suggesting that ATM loss contributes to tumorigenesis (Ref. Reference Choi, Kipps and Kurzrock34). Loss of ATM expression has also been observed in many cancer types including colorectal cancer, breast cancer, non-small cell lung cancer, lung adenocarcinoma and pancreatic cancer (Refs Reference Wang35–Reference Russell39). In addition to ATM mutation, ATM loss of heterozygosity (LOH) may arise through deletion of the long arm of chromosome 11 (11q). Interestingly, other DDR components are often co-deleted with ATM. Genes encoding MRE11, CHK1 and the histone variant H2AX are also located on chromosome 11q (Fig. 3), and are frequently deleted with ATM (Refs Reference Ouillette40, Reference Mlakar41). Allelic deletion of these genes may contribute to DNA damage repair deficiencies which could be targeted therapeutically. 11q deletion is commonly observed in breast cancer, chronic lymphocytic leukaemia (CLL), other lymphoid malignancies, and childhood neuroblastoma and is associated with poor survival (Refs Reference Mlakar41–Reference Monni and Knuutila43).
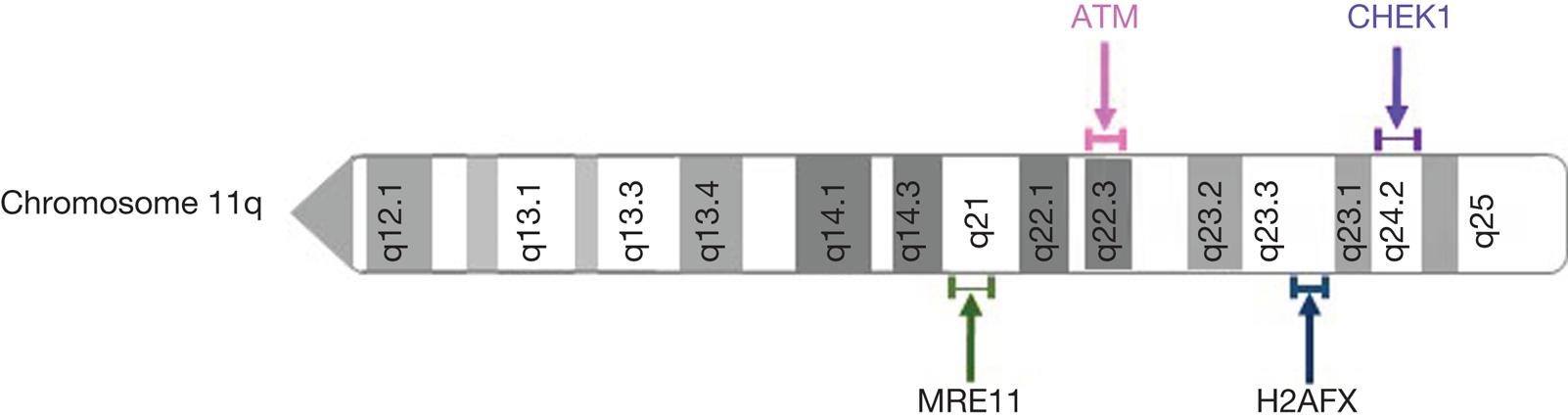
Fig. 3. Location of frequently deleted DNA damage response genes on chromosome 11q.
CHK2 mutations, although less common than mutations in ATM, are also observed across cancer types including colon, kidney, breast and prostate cancer (Ref. Reference Cybulski44). Homozygous germline CHK2 mutation is rare and manifests in Li-Fraumeni syndrome, a cancer predisposition syndrome usually associated with mutations in the gene encoding p53, TP53 (Ref. Reference Lee45). Somatic mutations in CHK2 have been observed across the entire amino acid sequence and lead to functionally null or unstable CHK2 protein.
The most commonly mutated gene across cancer types is TP53, which codes for the p53 protein. Around 80% of patients with Li-Fraumeni patients have germline mutations of TP53 (Ref. Reference Varley46). Somatic TP53 mutations are observed in around 40% of all tumours (Refs Reference Lawrence47, Reference Bouaoun48), the mutation rate varying between cancer types from nearly 100% in ovarian cancer to <10% for haematological malignancies (Ref. Reference Lawrence47). Mutations in TP53 result in a spectrum of p53 mutant proteins, from classical loss of transcriptional function to gain of function mutants which alter transcriptional networks and promote an oncogenic phenotype (Ref. Reference Hainaut and Pfeifer49).
As with ATM, TP53 loss of heterozygosity through allelic deletion of chromosome 17p is frequently observed in cancer (Ref. Reference Liu50). Loss of 17p is often accompanied by mutation of the other TP53 allele, although 17p deletion alone has been shown to predict poor prognosis in some myeloid malignancies (Refs Reference Fiskvik51, Reference Hallek52).
Overall aberration in the ATM/CHK2/p53 axis frequently occurs in cancer. Targeting cancer-specific defects in this pathway could contribute to effective cancer treatments with reduced side effects.
Rationale for the development of inhibitors
Neither ATM nor CHK2 kinases are essential for life, indicating some redundancy with other DNA damage signalling and repair pathways.
In humans, ATM mutations lead to the autosomal recessive disease A-T. A-T patients are very radiosensitive and display increased adverse and sometimes fatal reactions to both radiotherapy and radio-mimetic chemotherapy (Ref. Reference Rothblum-Oviatt33). In addition to the identification of ATM mutations in cancer (discussed in the section ‘Pathway dysfunction in cancer’), heterozygous carriers also have an increased risk of developing cancer, particularly breast and lymphoid (Refs Reference Choi, Kipps and Kurzrock34, Reference Bartek, Lukas and Bartkova53). ATM−/− mice are viable and display many features of A-T including cerebellar dysfunction, infertility, radio-sensitivity and cancer predisposition (Refs Reference Barlow54, Reference Xu and Baltimore55). Targeting ATM with small molecule inhibitors should sensitise cells to radio- and chemotherapy thus reducing the dose required and reducing off-target toxicities of these treatments. However, systemic ATM inhibition could also lead to increased toxicity from chemotherapy agents. For example, combining chemotherapy with other DDR inhibitors, such as MGMT and PARP inhibitors, led to dose reductions of both the chemotherapy and DDR inhibitors owing to increased toxicity (Refs Reference Jiang56, Reference Rabik, Njoku and Dolan57). Radiotherapy is more targeted towards the tumour and as techniques become more precise with improving technology. ATM inhibitors may be particularly useful in this context.
CHK2 kinase is also not essential. CHK2 knockout (KO) mice show little to no phenotype but, in contrast to ATM KO, are resistant to ionising radiation (IR) and have defects in p53-mediated apoptosis pathways (Ref. Reference Takai58). However, cancer-prone phenotypes associated with the absence of CHK2 become apparent when other DDR genes, such as CHK1, MRE11 and NBS1, are impaired (Ref. Reference Niida59). CHK1+/− CHK2−/− mice show high levels of spontaneous damage and decreased apoptotic responses, increasing cancer susceptibility showing some degree of co-operation and redundancy. Overall, the context in which CHK2 inhibitors will have therapeutic benefit remains unclear.
Preclinical development of ATM inhibitors
Many small-molecule ATP-competitive inhibitors of ATM have been developed and generally act as radiosensitisers in vitro (Ref. Reference Brandsma60). The first described potent and specific ATM inhibitor, KU55933, was developed by KuDos pharmaceuticals (now part of AstraZeneca) (Ref. Reference Hickson61). It enhanced the cytotoxicity of IR and topoisomerase I and II poisons, but its poor aqueous solubility and in vivo bio-availability precluded advanced preclinical testing. KU60019, a structural derivative of KU55933 with improved potency and aqueous solubility, effectively radiosensitised glioblastoma in vivo when directly injected into the tumour (Ref. Reference Biddlestone-Thorpe62), but still had poor in vivo bioavailability (Ref. Reference Weber and Ryan1). KU60019 caused greater radiosensitisation in p53 deficient tumours. However, using KU59403, another derivative of KU55933, in matched p53 functional and dysfunctional cell lines showed that the radio- and chemosensitising effects of ATM inhibition was not p53 dependent (Ref. Reference Batey63). While the pharmacodynamic properties of KU59403 were still not suitable for oral administration, systemic in vivo studies in mice were carried out by intraperitoneal injection. As well as sensitizing to IR, KU59403 also sensitised tumours to topoisomerase I and II inhibitors, irinotecan and etoposide respectively, in vivo.
AZ32 is a moderately potent ATM inhibitor discovered by chemical library screening at AstraZenena. The chemistry is different from that of the KuDos compounds and has been shown to be orally bioavailable as well as capable of crossing the blood-brain barrier in mice (Ref. Reference Karlin64). In vivo optimisation of AZ32 led to the development of AZD1390 sensitised brain tumours to radiotherapy in preclinical models justifying translation into a clinical trial (section ‘Clinical trials with ATM and CHK2 inhibitors’) (Ref. Reference Durant65).
In addition to AZD1390, the compound AZD0156 was developed by AstraZeneca following optimisation of a different lead scaffold (Ref. Reference Pike66). AZD0156 shows good pharmacodynamic properties and is synergistic with the topoisomerase I inhibitor irinotecan and the PARP inhibitor olaparib in tumour xenograft models. AZD0156 has also entered a phase 1 clinical trial.
CP-466722, another selective ATM inhibitor, was identified by Pfizer and showed similar radio-sensitising properties to KU55933 (Ref. Reference Rainey67). In vivo studies were not possible because of the compound having a short half-life in mice (t 1/2 < 1 hour).
In 2017, Dohmen et al. identified GSK635416A as a novel radio-sensitiser in non-small cell lung cancer (NSCLC) cell lines from a screen of published GlaxoSmithKline protein kinase inhibitors, which was shown to act through inhibition of ATM (Ref. Reference Dohmen68). When combined with the PARP inhibitor olaparib, GSK635416A showed an additive radio-sensitizing effect. No in vivo studies have been published for this compound to date.
Preclinical development of CHK2 inhibitors
In contrast to CHK1 inhibitors, few CHK2-specific inhibitors have been developed. In general, they show modest anti-proliferative effects when compared with ATM, ATR and CHK1 kinase inhibitors (Ref. Reference Ronco69).
A screen of the AstraZeneca compound library yielded AZD7762 as a potent CHK1 inhibitor with equal potency against CHK2 (Ref. Reference Zabludoff70). This dual inhibitor will be discussed with other CHK1 inhibitors in the section ‘Preclinical development of CHK1 inhibitors’.
A 2-arylbenzamidazole compound (ABI) was the first CHK2-specific inhibitor to be proposed, showing high selectivity (IC50 = 15 nM) over CHK1 (IC50 > 10 μM) (Ref. Reference Arienti71). However, CHK2 inhibition in cells by ABI was greatly reduced compared with cell-free assays achieving 42% inhibition of CHK2 at 5 μM. When used as a tool, the compound showed dose-dependent radioprotection in human CD4+ and CD8+ T-cells, similar to the radioresistance of CHK2 null mice.
Attenuation of IR-induced apoptosis was seen in mouse thymocytes after treatment with three other structurally distinct CHK2 inhibitors, VRX0466617 (Ref. Reference Carlessi72), PV1019 (Ref. Reference Jobson73) and CCT241533 (Ref. Reference Caldwell74). Another CHK2 inhibitor, BML-277 (CHK2 inhibitor II), first disclosed by Arienti et al. (Ref. Reference Arienti71), was shown to be radioprotective in human glioma cell lines (Ref. Reference Raso75). These data are consistent with the observation that CHK2 KO mice are radioresistant (section ‘Rationale for the development of inhibitors’ (Ref. Reference Takai58)). Studies in HT-29 (human colon cancer) cells and HeLa (human cervical cancer) cells treated with CCT241533 failed to show any impact on the radiomimetic bleomyocin cytotoxicity (Ref. Reference Anderson76). Interestingly, BML-277 antagonised oxaloplatin cytoxicity in colorectal cancer cell lines (Ref. Reference Pires, Ward and Dive77). In contrast, survival analysis by colony formation assay in U251 human glioblastoma cell line showed potentiation of IR by PV1019. Whether these contrasting observations reflect the differing molecular pathology of the cell lines remains to be determined.
Although the role of CHK2 inhibition in response to IR is unclear, there is some evidence that the combination of CHK2i with topoisomerase I poisons and poly (ADP)-ribose polymerase (PARP) inhibitors might be effective. PV1019 was shown to potentiate the cytotoxic effects of topotecan and camptothecin in ovarian cancer cell lines (Ref. Reference Jobson73). Potentiation of the effects of the PARP inhibitors rucaparib and olaparib was seen with the addition of CCT241533 (Ref. Reference Anderson76).
Clinical trials with ATM and CHK2 inhibitors
Three ATM inhibitors are currently being investigated in phase 1 clinical trials (Table 1). To date, no specific CHK2 inhibitor has progressed to clinical trials but phase 1 studies of AZD7762, the dual CHK1/CHK2 inhibitor have been undertaken in combination with gemcitabine. The results published from the completed trial showed that AZD7762 leads to cardiac toxicity leading to 2 further trials being terminated and the clinical progression of the inhibitor being discontinued (Ref. Reference Sausville78).
Table 1. ATM inhibitors currently in clinical trials

Role of ATR-CHK1-WEE1 in cell cycle checkpoints
The ATR-CHK1-WEE1 pathway activates both intra-S and G2/M checkpoint control in response to replication stress (RS) and DNA damage. RS is the momentary slowing or stalling of replication fork progression that can be caused by replication outstripping the rate of dNTP production or lesions in the DNA. ATR is activated by a number of DNA damaging factors, including ultraviolet radiation, antimetabolite-induced dNTP depletion, topoisomerase poisons, alkylating agents or DNA crosslinking agents (Ref. Reference Saldivar, Cortez and Cimprich79). Many of these factors result in single-strand DNA (ss-DNA), which allows for the recruitment of ATR activating proteins TOPBP1 or ETAA1. Although ss-DNA arises primarily from RS it is also a result of resected DSBs and nucleotide excision repair (NER) intermediates. Replication protein A (RPA) coats the ss-DNA, protecting it from degradation and enabling recruitment of ATR via ATR-interacting protein (ATRIP). ATR is activated by either TOPBP1, which is recruited by interaction with MRN complex that resects DSB to give long ss-DNA overhangs and the 9-1-1 complex (a proliferating cell nuclear antigen (PCNA)-like clamp that binds SS-DS DNA junctions) or by ETAA1, which is recruited to ss-DNA by interaction with RPA (Ref. Reference Thada and Cortez80). Upon activation, ATR activates CHK1 by phosphorylation at serine 345 causing CHK1 to autophosphorylate at serine 296, to achieve full activation. Like CHK2, CHK1 causes the inactivating phosphorylation of CDC25A/C (Refs Reference Rundle81, Reference Lewis82), thereby preventing them from removing the inhibitory phosphorylation on CDK2 and 1, respectively (as described in Fig. 4). In yeast and xenopus, CHK1 phosphorylates and activates Wee1 kinase activity to phosphorylate, and hence inactivate, CDK 1 and 2 (Refs Reference Lin, McNeely and Beckmann83, Reference Lee, Kumagai and Dunphy84). This is yet to be shown in mammalian cells. The net result of this kinase activation and phosphatase inhibition is inhibition of CDK2, and thus S-phase entry and progression, and inhibition of CDK1 preventing entry into mitosis.
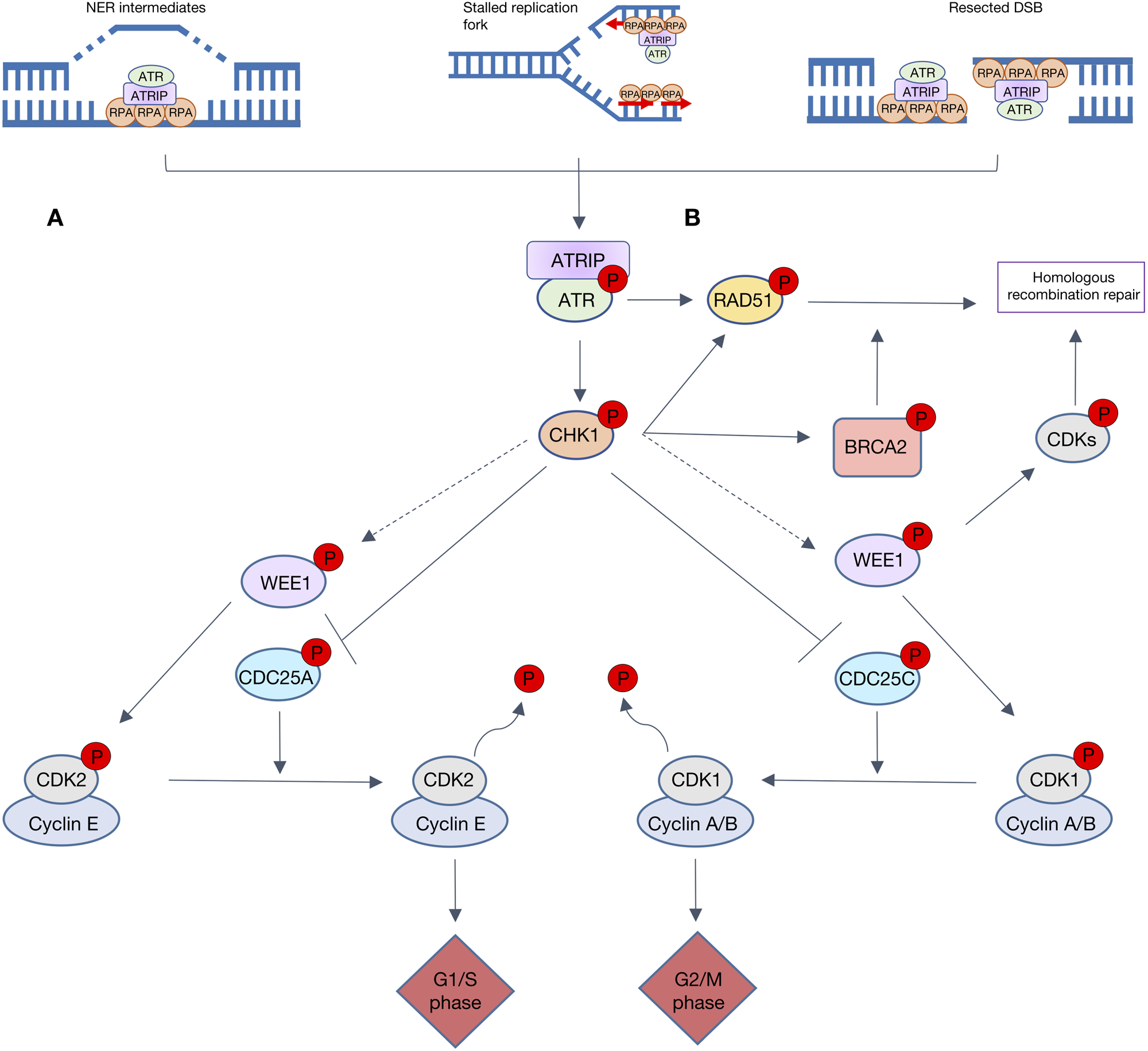
Fig. 4. Overview of ATR signalling in response to DNA damage. Resected double-strand breaks (DSBs), stalled replication forks and NER intermediates all lead to replication protein A (RPA) recruiting ATR via ATR-interacting protein (ATRIP). A. The ATR-CHK1 cascade is heavily involved in cell cycle checkpoint control. ATR activates CHK1 which causes the inactivating phosphorylation of both CDC25C and CDC25A, hence preventing the removal of inhibitory phosphorylation on CDK1 and 2, respectively. These lesions also activate WEE1, directly or via CHK1 (dashed grey arrows) to phosphorylate and inactivate CDK1 and 2. Progression through cell cycle G1/s and G2/M phases is reliant upon activation of CDK2/cyclin E and CDK1/cyclin A/B complexes, respectively. When WEE1 CHK1 or ATR are inhibited, CDK1 and CDK2 are activated so S-phase progression and mitotic entry occur with no delay to allow DNA repair. B. ATR, CHK1 and WEE1 also signal to key proteins involved in homologous recombination repair (HRR). ATR promotes repair protein RAD51 recruitment to DSBs and stalled replication forks, independent of BRCA. CHK1 phosphorylates key HRR proteins BRCA2 and RAD51. When activated WEE1 inhibitory phosphorylates CDKs, which play a key role in HRR end resection.
Role in DNA repair
The ATR-CHK1-WEE1 cascade also plays a role in HRR, a high fidelity DSB repair pathway, restricted to late S and G2 phase as it uses the sister chromatid as a DNA template (Ref. Reference Wyman, Ristic and Kanaar85). During HRR extensive DNA end-resection occurs, resulting in the ss-DNA overhang that leads to ATR activation. HRR is also responsible for the resolution of collapsed replication forks caused by RS – the prime activator of the ATR-CHK1-WEE1 pathway. It is therefore not surprising that these kinases are also associated with DNA repair by HRR (Ref. Reference Brown86).
All three kinases in the cascade have demonstrated involvement in HRR. In BRCA defective cells, ATR can act independently of BRCA1 to recruit RAD51 to DSBs and stalled replication forks, inhibition of ATR disrupted RAD51 loading suggesting key involvement in HRR (Ref. Reference Yazinski87). Similarly, CHK1 may promote HRR by phosphorylating key HRR components BRCA2 and RAD51 (Refs Reference Rundle81, Reference Sørensen88). WEE1 is also involved in HRR owing to its inhibitory phosphorylation of CDKs upon activation. The resection of DNA ends, a necessary step in HRR is antagonised by CDK activity (Ref. Reference Buisson89), inhibition of CDK activity by WEE1 (or cdc25 inactivation downstream of CHK1) promotes HRR, therefore inhibition of either CHK1 or WEE1 will result in higher CDK activity and compromise HRR (Ref. Reference Krajewska90).
Pathway dysfunction in cancer
The importance of the ATR-CHK1-WEE1 cascade is highlighted by the embryonic lethality of all three components (Refs Reference de Klein91–Reference Tominaga93). No humans are recorded as being born without these essential kinases but Seckel syndrome (SS) is a rare, autosomal recessive disorder owing to a hypomorphic mutation in ATR, resulting in delayed development but not cancer predisposition. SS mice are not cancer-prone, even when crossed with p53 defective mice (Ref. Reference Murga94). There are contrasting data regarding the tumour predisposition of ATR+/− mice with one study reporting increased tumour incidence and others reporting no increase in tumour incidence (Refs Reference de Klein91, Reference Brown and Baltimore95). CHK1+/− mice are not predisposed to tumourigenesis (Ref. Reference Liu92), and no abnormalities are reported in adult WEE1+/− mice (Ref. Reference Tominaga93).
The general consensus seems to be that complete loss of ATR, CHK1 or WEE1 signalling is incompatible with normal development but that compromising the pathway by hypomorphic mutation or heterozygous deletion does not predispose to tumour development. However, upregulation of the pathway in tumours may be indicators of poor prognosis. Two studies in breast cancer indicate that high pCHK1 levels correlated with local recurrence and worse cancer-specific survival (Refs Reference Macheret and Halazonetis96, Reference Magdalou97) and WEE1 overexpression have been observed in several tumour types: hepatocellular carcinoma (HCC), breast, glioblastoma, lung and colon (Ref. Reference Berti and Vindigni98).
Rationale for targeting ATR-CHK1-WEE1
Cancer cells are considered to have higher levels of RS than normal cells. There are several causes for this: (i) increased expression of oncogenes or growth factor receptors that drive cells into S-phase, (ii) accelerated cell cycle progression owing to increased expression of CDKs or their cyclin partners or loss of their protein inhibitors, and (iii) loss of G1 checkpoint control (Refs Reference Macheret and Halazonetis96–Reference Massague99). Additionally, RS results in genomic instability that is an enabling characteristic of cancer (Refs Reference Kotsantis, Petermann and Boulton100, Reference Hanahan and Weinberg101), thereby creating a vicious circle. RS is the prime trigger for ATR-CHK1-WEE1 signalling and cancer cells are therefore highly dependent on this pathway (Refs Reference Kotsantis, Petermann and Boulton100, Reference Syljuåsen102, Reference Kitao103). Thus, there is a potential to exploit the increased RS, coupled with the loss of G1 control, in cancer cells by targeting the ATR-CHK1-WEE1 pathway, without compromising normal cells with proficient G1 checkpoint control (Ref. Reference Kim104).
In addition to their pivotal role in the S and G2/M cell cycle checkpoints, ATR-CHK1-WEE1 also promote HRR, as described above. Therefore, inhibiting these kinases has the potential to compromise HRR, thereby sensitising cells to DNA damaging anticancer agents.
Preclinical development of ATR inhibitors
An early study in 1998 found that overexpression of kinase-inactive ATR caused sensitivity to IR, cisplatin and methyl methanesulfonate (MMS) (Ref. Reference Cliby105). Caffeine was found to be a weak ATR inhibitor (1999) and, although it lacked specificity, it was still good enough to test the potential of ATR inhibition (Ref. Reference Hall-Jackson106). It was shown to inhibit ATR activity at a radio sensitising concentration (Ref. Reference Sarkaria107). Subsequently in 2002, Nghiem et al., showed that expression of the kinase-dead ATR conferred sensitivity to multiple anti-cancer/DNA damaging agents (UV, hydroxyurea (HU), IR, cisplatin and aphidicolin). In terms of the potential of single-agent ATR inhibitors this study also showed that endogenous causes of replication stress (cyclin D, E, CDK2 overexpression or p53 inactivation by MDM2 or human papillomavirus (HPV) E6 expression) conferred sensitivity to kinase-dead ATR overexpression and/or caffeine (Ref. Reference Nghiem108).
ATR inhibitor development was slow to take off from these early studies, possibly because of the difficulty in developing a cell-free assay. Nevertheless, in 2011, NU6027, originally developed as a CDK2 inhibitor, was found to be a more potent inhibitor of ATR that CDK2 in intact cells. NU6027 enhanced cisplatin and HU cytotoxicity in an ATR-dependent manner, and the major classes of DNA damaging anticancer drugs in MCF7 breast cancer cells, and attenuated G2 cell cycle arrest. Cells defective in HRR are exquisitely sensitive to PARP inhibitors and, in the first investigation of its kind, NU6027 inhibited HRR and increased PARP inhibitor cytotoxicity (Ref. Reference Peasland109).
In 2011 a novel ATRi screen identified ETP-46464 as an ATRi that had increased cytotoxicity in cells overexpressing cyclin E. It significantly sensitised cells to IR, abolishing the G2/M checkpoint in these cells, independent of p53 status (Ref. Reference Toledo110). In 2015, Teng et al., went on to show that it also sensitised cells to cisplatin treatment (Ref. Reference Teng111). However, ETP-46464 lacked specificity as it also inhibited mTOR, DNA-PKcs and P13Kα, and had poor in vivo pharmacological properties (Ref. Reference Toledo110).
AZ20 is an ATRi developed in 2013 from the P13K inhibitor, LY294002, with good potency and selectivity (Ref. Reference Foote112). In acute myeloid leukaemia (AML) cell lines and patient samples, AZ20 acted synergistically with cytarabine, resulting in enhanced apoptosis and induced replication stress (Ref. Reference Ma113). This drug also synergistically inhibited cell growth in combination with gemcitabine in pancreatic cancer cell lines (Ref. Reference Liu114).
AZD6738 was developed from AZ20 in 2018, with improved aqueous solubility and excellent pharmacokinetic qualities (Ref. Reference Foote115). It sensitised non-small cell lung cancer (NSCLC) cells and xenografts to cisplatin and gemcitabine (Ref. Reference Vendetti116), and a panel of human cancer cells to radiation (Ref. Reference Dillon117). AZD6738 suppressed tumour growth and increased apoptosis in ATM defective cells (Ref. Reference Min118).
VE-821 was one of the first potent ATRi with greatly improved selectivity for ATR over other PI3K-like kinases discovered in 2011 (ATM, DNAPKcs, mTOR). VE-821, as a single agent, increased apoptosis in cancer cells versus non-cancerous cells (Ref. Reference Reaper119). VE-821 can sensitise cells to IR (Refs Reference Alsubhi120–Reference Šalovská122), gemcitabine and camptothecin (Refs Reference Reaper119, Reference Jossé123). However, the strongest synergy observed thus far is with platinum-based therapies cisplatin and carboplatin (Refs Reference Reaper119, Reference Huntoon124). Interestingly, when used in combination with the PARPi veliparib, VE-821 further sensitised BRCA defective cells beyond the sensitivity already observed owing to HRR status (Ref. Reference Huntoon124). In 2015 Middleton et al., showed that defects in ATM, HRR (BRCA2, XRCC3) and BER (XRCC1) resulted in increased sensitivity to VE-821. Interestingly, defective Ku80 (involved in NHEJ) caused hypersensitivity to VE-821, but the loss of its binding subunit, DNA-PKcs, did not (Ref. Reference Middleton125).
M6620 (also known as VE-822/VX-970 developed in 2012 and, from the same chemical series as VE-821) was the first highly selective, potent ATR inhibitor to go into clinical trials and is currently in phase 2 trials. It potentiates a number of DNA damaging agents including carboplatin, cisplatin, gemcitabine, irinotecan and IR in a wide array of cancers (Refs Reference Hall126–Reference Nagel132). Nagel et al., found that M6620 combined with cisplatin showed a better response in vivo than cisplatin combined with etoposide, another chemotherapeutic, providing a solid rationale for combining cisplatin and M6620 in the clinic and limiting inevitable side effects with combining two chemotherapeutic agents (Ref. Reference Nagel132). As with other ATR inhibitors, it was found that ATM conferred sensitivity to ATR, both in vitro and in vivo (Refs Reference Shi128, Reference Schmitt133).
In 2017 Wengner et al., characterised a novel ATRi, BAY1895344, which inhibited cell proliferation in an array of human cancer cell lines as well as having a strong anti-tumour effect as monotherapy in xenograft models. Synergistic anti-cancer activity was reported when used in combination with Radium-223 in xenograft models (Ref. Reference Wengner134).
Preclinical development of CHK1 inhibitors
UCN-01 was a first-generation, potent CHK1 inhibitor originally developed in 1999 as a protein kinase C inhibitor (Ref. Reference Shapiro and Harper135). In vitro, it abrogated G2 checkpoint control and sensitised p53 defective cancer cells to DNA damaging agents (cisplatin, camptothecin and IR). However, it has poor potency and specificity and struggled to bypass a radiation-induced G2/M checkpoint (Ref. Reference Graves136).
In 2008 the biological effects of AZD7762, a potent dual CHK1 CHK2 inhibitor with equal potency against both kinases, was shown to result primarily from inhibition of CHK1 (Ref. Reference Zabludoff70). In vitro studies have demonstrated that AZD7762 potentiated the cytotoxic effects of the nucleoside analogue gemcitabine, topoisomerase inhibitors and cisplatin (Refs Reference Xiao137–Reference Bartucci141). These findings were reflected in vivo and AZD7762 showed good pharmacokinetics and tolerability in mice. However, the effect of dual CHK1/CHK2 inhibition showed no increased benefit compared with CHK1-specific targeting agents suggesting that most of the anti-tumour effects are through inhibition of CHK1.
PF-477736 is a potent ATP-competitive CHK1 inhibitor with >100-fold selectivity over CHK2 developed in 2008 (Ref. Reference Blasina142). It has potent single-agent activity in triple-negative breast and ovarian cancer cell lines (Ref. Reference Bryant, Rawlinson and Massey143), as well as sensitising cells to chemotherapeutic drugs gemcitabine, carboplatin, doxorubicin, mitomycin C and toptotecan (Refs Reference Blasina142, Reference Kim, James and Annunziata144–Reference Zhang146). PF-477736 also sensitises HPV positive head and neck cancer cells to radiation (Ref. Reference Busch147). PF-477736 caused synergistic cytotoxicity in combination with targeted therapies irutinib and bosutinib in mantle cell lymphoma (MCL) and chronic myeloid leukaemia (CML), respectively (Refs Reference Restelli148, Reference Nguyen149). PF-477736 was more cytotoxic in p53 mutant and Myc-driven cancers (Refs Reference Blasina142, Reference Ferrao150).
In 2017, a novel CHK1 inhibitor, MK-8776 (also known as SCH900776 and identified in 2011), abrogated IR-induced G2/M checkpoint activation, resulting in aberrant mitosis, and was a potent radiosensitiser in breast and cervical cell lines (Refs Reference Suzuki151, Reference Zhou152). MK-8776 also radiosensitised non-small cell lung cancer and head and neck cancer cell lines, in p53 non-functional cells (Ref. Reference Bridges153). Montano et al., reported that MK-8776 sensitised cells to an array of DNA damaging agents: HU (20–70 fold), cytarabine (15–35 fold) and gemcitabine (5–10 fold), with no sensitisation reported with cisplatin or 5-fluorouracil (5-FU) (Ref. Reference Montano154). However, a later study by Herudkova et al. found that MK-8776 significantly sensitised cells to cisplatin and another platinum-based therapy, LA-12 (Ref. Reference Herůdková155).
SRA737 (previously CCT244747, which was discovered in 2012) is a novel, potent, orally active CHK1 inhibitor, with good selectivity (Refs Reference Walton156, Reference Patel157). It was developed at the ICR and is active as a single agent in MYCN-driven neuroblastoma and in combination with IR, gemcitabine and irinotecan (Refs Reference Walton156, Reference Patel157). SRA737 has synergistic antitumour activity with the PARP inhibitors niraparib and olaparib in mammary and ovarian cancer cells in vitro and in vivo (Ref. Reference Booth158).
In 2015 LY2606368, a specific CHK1 inhibitor with strong single-agent activity in vitro and in vivo was discovered (Refs Reference King159–Reference Lowery162). It demonstrated synergy with PARP inhibitors olaparib and BMN673 in ovarian and gastric cancer, respectively (Refs Reference Brill163, Reference Yin164) and potentiated cisplatin even in a panel of platinum-resistant human cancers cell lines (Refs Reference Sen161, Reference Hsu165).
Pre-clinical development of WEE1i
Despite the key role for WEE1 in S and G2 arrest, very few small-molecule inhibitors have been developed. In 2001, PD0166285 was the first potent WEE1 inhibitor. It radiosensitised ovarian, colon, lung and ovarian tumour cells in a p53-dependent manner (Ref. Reference Wang166). It was a potent radiosensitiser in glioblastoma, but a major limitation to its development is its inability to penetrate the blood-brain-barrier (Refs Reference Mir167–Reference de Gooijer169). Furthermore, PD0166285 was non-selective, it also inhibited CHK1, Src non-receptor tyrosine kinase, epidermal growth factor receptor (EGFR), platelet-derived growth factor β (PDGF-β) and fibroblast growth factor receptor-1 (FGFR-1) (Ref. Reference Panek170).
In 2009 AZD1775/MK-1775 was discovered as a specific, potent WEE1 inhibitor that showed excellent selectivity (Ref. Reference Hirai171). It was effective as a single agent (Ref. Reference Kreahling172), as well as in synergistically increasing the cytotoxicity of various DNA damaging agents (IR, gemcitabine, carboplatin, cisplatin, 5-FU, pemetrexed, doxorubicin and camptothecin), in vitro and in vivo (Refs Reference Hirai171, Reference Hirai173–Reference Sarcar176). Earlier studies reported p53 status as a determinant for sensitivity to AZD1775 (Refs Reference Hirai171, Reference Rajeshkumar174). However, more recent studies have shown MK-1775 cytotoxicity to be independent of p53 (Refs Reference Kreahling172, Reference Heijink177). Heijink et al. carried out a genome-wide unbiased screen and concluded that the activity of DNA replication proteins, beyond p53, is a key determinant of WEE1 inhibitor sensitivity (Ref. Reference Heijink177).
Synergy between inhibitors of ATR-CHK1-WEE1
Studies of the combination of inhibitors of the pathway with one another have thus far shown potential. ATR inhibition by VE-821 and CHK1 inhibition by AZD7762 caused synergistic cell death in vitro and in vivo (Ref. Reference Sanjiv178). VX-970 (VE-822) was well tolerated in combination with AZD7762 in mice and resulted in increased survival of tumour-bearing mice (Ref. Reference Sanjiv178). ATR inhibition by AZD6738 was also synergistic with WEE1 inhibition by AZD1775 in causing the accumulation of DNA damage, via forced mitotic entry, and growth inhibition. This combination also inactivated HRR- sensitising cells to cisplatin and PARP inhibition (Ref. Reference Jin179). An anti-metastatic effect was observed in vitro when ATR inhibitors AZ06738 and ETP-46464 were combined with WEE1 inhibitor AZD1775 (Ref. Reference Bukhari180).
CHK1 and WEE1 inhibitors are another well-tolerated, synergistic combination as demonstrated in several studies in a variety of different cancer models, a few examples are given here. CHK1 inhibitor MK-8776 sensitised AML cells to AZD1775 ex-vivo and the combination was effective against neuroblastoma xenografts (Refs Reference Chaudhuri181, Reference Russell182). A study by Hauge et al., showed synergistic anti-tumour effects between the WEE1 inhibitor AZD1775 and CHK1 inhibitors AZD7762 and LY2603618. A combination of these inhibitors resulted in mitotic catastrophe and reduced cell survival because of the combined effects on S phase and DNA damage associated with unscheduled replication initiation (Ref. Reference Hauge183). Similarly, the combination of CHK1 inhibitor AZD7762 and WEE1 inhibitor AZD1775 caused increased cytotoxicity and apoptosis in metastatic melanoma cell lines (Ref. Reference Magnussen184). Synergy was also reported with CHK1 inhibitor PF-477736 and WEE1 inhibitor AZD1775 in MCL cells (Ref. Reference Chilà185).
Pre-clinical synergy of ATR-CHK1-WEE1 inhibitors and PARP inhibitors
Inhibitors of the ATR-CHK1-WEE1 cascade have shown synergy with PARP inhibitors. Peasland et al., were the first to show that the ATR inhibitor, NU6027, was synthetically lethal in combination with the PARP inhibitor, rucaparib in breast and ovarian cancer cells (Ref. Reference Peasland109). Subsequently, the ATR inhibitor VE-821 was found to sensitise BRCA mutant cells to veliparib (Ref. Reference Middleton125) and a synthetically lethal screen found VE-821 had profound synergy with PARP inhibition in both HRR competent and defective cells (Ref. Reference Mohni186). Kim et al., showed that inhibiting PARP resulted in increased reliance on the ATR-CHK1 pathway for genomic stability and that the combination of olaparib with ATRi AZD6738 effectively reduced tumour burden in patient-derived xenografts of serous ovarian cancer (Ref. Reference Kim187).
A series of PARP inhibitors (rucaparib, olaparib, veliparib and NU1025) synergised with various CHK1 inhibitors (UCN-01, AZD7762 and LY2603618) to increase DNA damage and apoptosis in vitro in breast cancer cells (Ref. Reference Booth188). The PARPi olaparib in combination with CHK1 inhibitor MK-8776, supressed colony formation in BRCA mutant models to a greater degree than either inhibitor as a single agent (Ref. Reference Kim187). The PARP inhibitor Talazoparib was also synergistic with LY2606368, both in vitro and in vivo in gastric cancer (Ref. Reference Yin164). Similarly, LY2606368 also showed inhibited HRR function and synergised with olaparib to decrease cell survival in BRCA wild-type cells (Ref. Reference Brill163).
Fewer studies have looked at PARP and WEE1 inhibition combined, perhaps owing to the availability of only one WEE1 inhibitor with the desired selectivity and specificity (AZD1775). AZD1775 has only been tested in combination with olaparib but these results have been promising and when used in combination, the inhibitors act synergistically to radiosensitise pancreatic, and KRAS-mutant NSCLC cells further than when either is used as a single agent (Refs Reference Karnak189, Reference Parsels190).
ATR inhibitors in clinical trials
M6620 (VE-822/VX-970) was the first ATR inhibitor to reach clinical trials. Thomas et al., first reported ATR inhibition in combination with chemotherapy in patients, with the maximum dose of topotecan being well tolerated when used in combination with M6620 (Ref. Reference Thomas191). There are currently three active clinical trials using M6620, one of which is the first in a human study looking at the pharmacokinetics of M6620 in combination with gemcitabine, cisplatin, etoposide carboplatin and irinotecan (NCT02157792). A number of phase 1 and 2 studies are currently recruiting patients for the use of M6620 single agent and in combination with a number of DNA damaging agents including irradiation, cisplatin, carboplatin, gemcitabine, irinotecan and topotecan (see Table 2). Another selective, bioavailable ATR inhibitor currently in phase 1 and 2 trials is AZD6738. Of these trials, nine are investigating the use of AZD6738 with the PARP inhibitor olaparib, which has strong support from pre-clinical data (Ref. Reference Kim187). The ATR inhibitor BAY1895344 is in a first in human phase I safety trial in patients with advanced solid tumours and lymphomas (NCT03188965). M4344/VX-803 is an orally bioavailable ATR inhibitor currently recruiting in phase 1 clinical trial where it will be used as a monotherapy and in combination with cisplatin, carboplatin or gemcitabine (NCT02278250). The ATR inhibitor BAY1895344 has recently shown to have anti-tumour activity and is well tolerated at active doses in cancers with defects in DDR, such as loss of ATM (Ref. Reference Bono192) (NCT03188965).
Table 2. ATR inhibitors currently in clinical trials

CHK1 inhibitors in clinical trials
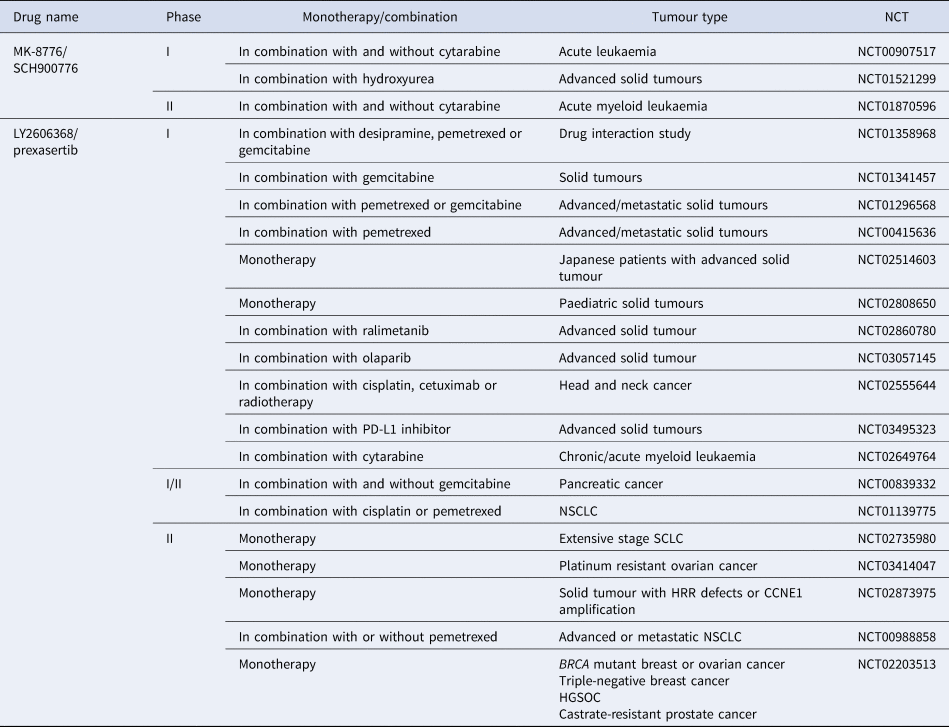
Although there is significant interest surrounding CHK1 inhibitors, clinical progression has often been hindered because of the lack of bio-availability and off-target effects (Ref. Reference Patel157). UCN-01 was used in combination with carboplatin in a phase 1 study (NCT00036777) and progressed into phase 2 clinical trials in 2010 in patients with metastatic melanoma, but the trial was terminated prematurely because of discouraging results (NCT00072189). CHK1 inhibitors with greater specificity have now entered clinical evaluation. SRA737 is currently in phase 1/2 clinical trials, both as a monotherapy and in combination with gemcitabine +/− cisplatin (NCT02797964) (NCT02797977). MK-8776 is also in phase 1 and 2 trials as a monotherapy and in combination with gemcitabine, cytarabine and hydroxyurea (NCT00779584) (NCT01870596). Of the four clinical trials it is currently in, two have been completed, with one terminated and one withdrawn owing to a lack of patients (see Table 3). However, in vitro studies have shown MK-8776 to have a short half-life, as well as undergoing rapid demethylation in vivo, resulting in a less selective metabolite (MU379) (Ref. Reference Samadder193). Phase 1 clinical trials, in patients with advanced solid tumours, of PF-477736 in combination with gemcitabine were terminated early for business reasons, rather than safety concerns (NCT00437203). Prexasertib (LY2606368) is currently in phase 2 trials a monotherapy agent, specifically in patients with cancers that are p53 mutant, have DDR defects such as BRCA mutation, increased replication stress or CCNE1 amplification, as these are determinants of CHK1 inhibitor sensitivity (NCT02735980, NCT02203513, NCT03414047, NCT02873975). Prexasertib has also entered phase 1 and 2 clinical trials in combination with pemetrexed (NCT01296568, NCT00415636, NCT01139775, NCT00988858), gemcitabine (NCT01358968, NCT01341457, NCT01296568, NCT00839332) or cisplatin (NCT02555644, NCT01139775) but has currently only been in one clinical trial with a PARP inhibitor (NCT03057145), despite promising synergy being observed pre-clinically (Refs Reference Brill163, Reference Yin164).
Table 3. CHK1 inhibitors currently in clinical trials
WEE1 inhibitors in clinical trials
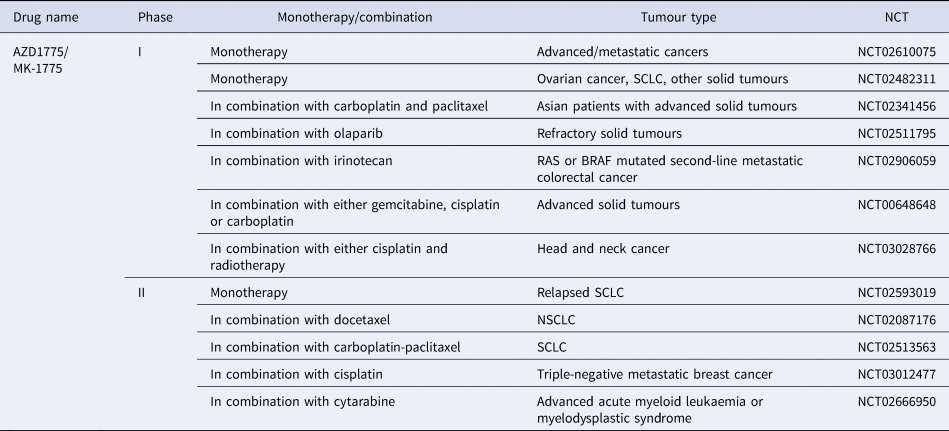
AZD17775 is the only WEE1 inhibitor to reach clinical development and is already in a number of phase 1 and 2 trials being used in combination with treatments such as carboplatin, gemcitabine, cisplatin, cytarabine and olaparib (Refs Reference Leijen194, Reference Oza195, Reference Lheureux196). Recently a dose-escalation trial of AZD1775 in combination with gemcitabine and radiation showed promising results with good tolerability in patients with locally advanced pancreatic cancer (Ref. Reference Cuneo197). The majority of clinical trials are still recruiting, suggesting this kinase inhibitor has exciting potential once trials have been completed and data is collected and analysed (Table 4).
Table 4. WEE1 inhibitors currently in clinical trials
DNA checkpoint kinase and immune checkpoint inhibitor combinations
Immune checkpoint inhibitors block the immunosuppressive mechanisms employed by cancers to prevent an effective anti-tumour immune response and have been found to be efficacious in many types of cancer. There is increasing evidence that tumour mutational burden increases the immunogenicity of cancers through the production of mutation-associated neoantigens, including those associated with microsatellite instability from defective DNA mismatch repair (Ref. Reference Bever and Le198). Damaged cytosolic DNA may also directly activate the immune system by stimulating interferon via the STING pathway (Stimulation of Interferon Genes) leading to enhanced immune checkpoint inhibitor responses in pre-clinical models. In ATM-deficient mice and patients with ataxia telangiectasia enhanced interferon production through the STING pathway has been observed (Ref. Reference Bever and Le198). ATM inhibition has recently been found to increase type 1 interferon signalling in a STING independent manner (Ref. Reference Zhang199). Clinical trials evaluating DNA damage repair inhibitors with immune checkpoint inhibitors are ongoing, including PARP, ATR and CHK1 inhibitors (Table 2).
Concluding remarks and future directions
Targeting DDR checkpoint signalling has evolved to the clinic based on sound scientific hypotheses and preclinical data. Early less specific inhibitors may have clouded the case for development but now more specific inhibitors are under investigation both as monotherapy and in combination with conventional cytotoxic chemotherapy or novel agents. The preclinical data suggest that targeting the ATR-CHK1-WEE1 pathway is likely to be more fruitful than targeting ATM and CHK2 signalling. To date, the most active combinations for each class of kinase inhibitor include ATR inhibitors with cis/carboplatin and CHK1 inhibitors with gemcitabine. A developing field is a potentiation with immune checkpoint inhibitors via several mechanisms of action. Identification of predictive biomarkers, particularly for monotherapy, however, has been challenging, for example, whether the presence of TP53 mutations confers sensitivity.
The potential for some of these agents to be associated with second malignancy must not be forgotten, which is particularly a concern for young patients treated with these agents. Since defects in ATM and CHK2 are associated with tumours, but defects in ATR, CHK1 and WEE1 are not, one might predict that as single agents, inhibitors of the former might be associated with second malignancies but not the latter. However, in combination with cytotoxics already associated with second malignancies, the incidence is likely to be increased unless lower doses of the primary cytotoxic can be used in combination to achieve the same efficacy. Nevertheless, we must remember that the malignancies for which these agents are most likely to be used, especially in children, are the ones that are most difficult to cure with current strategies in which cure has yet to be achieved for the majority at any cost rather than at least cost.
Acknowledgements
This work was partly funded by the Children's Cancer & Leukaemia Group and Little Princess Trust and the JGWP foundation.








