


Inherited neurotransmitter disorders affecting monoamine, tetrahydrobiopterin and γ amino butyric acid (GABA) metabolisms are rare disorders. Catecholamine (dopamine, epinephrine and norepinephrine) and serotonin metabolism disorders include tyrosine hydroxylase (TH) (OMIM# 605407) (EC 1.14.16.2) and aromatic l-amino acid decarboxylase (AADC) (OMIM# 608643) (EC 4.1.1.28) and biopterin metabolism disorders include guanosine triphosphate cyclohydrolase I (OMIM# 128230) (EC 3.5.4.16), 6-pyruvoyltetrahydropterin synthase (PTPS) (OMIM# 261640) (EC 4.2.3.12), sepiapterin reductase (OMIM# 612716) (EC 1.1.1.153) and dihydropteridine reductase (DHPR) (OMIM# 261630) (EC 1.5.1.34) deficiencies.Reference Pearl 1 - Reference Opladen, Hoffmann and Blau 3 Positive newborn screening for phenylketonuria and tetrahydrobiopterin loading test leading to normalization of plasma phenylalanine levels within 24 hours are diagnostic for PTPS, DHPR and pterin-4a-carbinolamine dehydratase deficiencies.Reference Pearl 1 - Reference Opladen, Hoffmann and Blau 3 Clinical features are usually infantile onset and include tremor, hypokinesia, rigidity, dystonia, chorea, motor and cognitive dysfunction, failure-to-thrive, sleep disturbances and autonomic dysfunction (e.g., blood pressure fluctuations, abdominal distention, temperature instability, constipation and diarrhea episodes and fatigue) secondary to catecholamine and serotonin deficiency.Reference Pearl 1 - Reference Hyland 4 Measurement of CSF neurotransmitter metabolites, including homovanillic acid (HVA), 5-hydroxyindolacetic acid (5-HIAA), 3-O-methyldopa (3-OMD), tetrahydrobiopterin, biopterin and neopterin, is a useful diagnostic tool to identify disorders of catecholamine, serotonin and biopterin metabolisms.Reference Pearl 1 , Reference Hyland 4 The diagnosis is confirmed by direct sequencing of candidate genes based on the CSF neurotransmitter metabolite profiles suggestive of a specific enzyme deficiency.Reference Pearl 1 - Reference Hyland 4
γ Amino butyric acid metabolism defects are succinic semialdehyde dehydrogenase (SSADH) (OMIM# 271980) and GABA transaminase (OMIM# 613163) (EC 2.6.1.19) deficiencies.Reference Pearl, Wiwattanadittakul, Roullet and Gibson 2 Neonatal-onset seizures are clinical features of GABA transaminase deficiency. Infantile-onset hypotonia, global developmental delay and/or seizures are clinical features of SSADH deficiency. Diagnosis of SSADH deficiency is suspected by elevated 4-hydroxybutyrate in urine organic acid analysis, whereas GABA transaminase deficiency is suspected by elevated GABA and β-alanine levels in CSF amino acid analysis.Reference Pearl, Wiwattanadittakul, Roullet and Gibson 2 The diagnosis is confirmed by direct sequencing of candidate genes.
In this study, we report outcome of patients with pediatric inherited neurotransmitter disorders affecting monoamine, tetrahydrobiopterin and GABA metabolisms from a single Inherited Neurotransmitter Disorder Clinic to increase our knowledge regarding those rare disorders.
This study was approved by the Institutional Research Ethics Board (Approval# 1000057003). All patients with disorders of monoamine, tetrahydrobiopterin and GABA metabolisms seen in our Inherited Neurotransmitter Disorder Clinic were included. All patients were referred for the management and treatment after their suspected diagnosis. We reviewed electronic patient charts for clinical features, neuroimaging, biochemical investigations, molecular genetic investigations and treatment. Targeted direct sequencing of relevant genes was performed in various clinical molecular genetics laboratories according to their methods.
In total, 12 patients with inherited neurotransmitter disorders affecting monoamine, tetrahydrobiopterin and GABA metabolisms diagnosed during January 1999 and March 2017 were included. Demographics, clinical features, biochemical and molecular genetic data for all patients are listed in Table 1a. Three patients had primary disorders of monoamine metabolism including TH deficiency (n=2) (one patient reported previouslyReference Mercimek-Mahmutoglu, Sidky and Hyland 5 ) and AADC deficiency (n=1). Seven patients had disorders of tetrahydrobiopterin metabolism including PTPS deficiency (n=4) (one patient reported previouslyReference Mercimek-Mahmutoglu, Sidky and Hyland 5 ) and DHPR deficiency (n=3) (one patient reported previouslyReference Mercimek-Mahmutoglu, Sidky and Hyland 5 ). Six out of these seven patients were identified by positive newborn screening for phenylketonuria. Owing to normalization of phenylalanine levels after tetrahydrobiopterin loading test, further investigations including elevated urine neopterin (biomarker for PTPS deficiency) or deficient DHPR activity in blood dot spot confirmed the biochemical diagnosis in the neonatal period in six patients. Their genetic diagnosis was confirmed by direct sequencing of PTS (for PTPS deficiency) and QDPR (for DHPR deficiency), respectively. One patient with DHPR deficiency was initially treated as if the disorder was phenylketonuria. The correct diagnosis of DHPR deficiency was made at 11 months of age when the child had global developmental delay and seizures. Two patients were diagnosed by low CSF HVA suggestive of TH deficiency. One patient was diagnosed by low HVA and 5-HIAA and high 3-OMD suggestive of AADC deficiency. Genetic diagnosis was confirmed by direct sequencing of TH in two patients. One patient with AADC deficiency underwent whole-exome sequencing in addition to CSF neurotransmitter analysis requested at the same time, confirming the genetic diagnosis.
Table 1a Clinical features, neuroimaging, biochemical and molecular genetics results of patients with inherited metabolic disorders presenting with inherited neurotransmitter disorders

↑=elevated; ↓=decreased; AADC=aromatic l-amino acid decarboxylase; d=day(s); DHPR=dihydropteridine reductase; FTT=failure to thrive; GB=globus pallidus; GDD=global developmental delay; GLT=glutamate; GLU=glutamine; 4-HBA=4-hydroxybutyric acid; 5-HIAA=5-hydroxyindol acetic acid; HVA=homovanillic acid; m=month(s); IUGR=intrauterine growth retardation; MRS=magnetic resonance spectroscopy; N=normal; NA=not available; NeP=neopterine; NP=not performed; 3-OMD=3-O-methyldopa; Phe=phenylalanine; PTPS=6-pyruvoyltetrahydropterin synthase; RBC=red blood cell; SSADH=succinic semialdehyde dehydrogenase; THB=tetrahydrobiopterine; TH=tyrosine hydroxylase; y=year(s).
* Fathers are brothers.
** Parents are first cousins.
Age-appropriate reference ranges for biochemical investigations: Plasma phenylalanine 45-65 µmol/L; urine neopterin/biopterin ratio 0.2-3.0; blood dot spot DHPR activity 7-22 µmol/g hb; AADC RBC activity 36-129 pmol/minute/ml.
CSF neurotransmitters age-appropriate reference range: CSF HVA: 0-0.2 years=337-1299 nmol/L; 0.2-0.5 years=450-1132 nmol/L; 0.5-2 years=294-1115 nmol/L; 2-5 years=233-928 nmol/L; 5-10 years=218-852 nmol/L; 10-15 years=167-563 nmol/L; adults=145-324 nmol/L.
CSF 5HIAA: 0-0.2 years=208-1159 nmol/L; 0.2-0.5 years=179-711 nmol/L; 0.5-2 years=129-520 nmol/L; 2-5 years=74-345 nmol/L; 5-10 years=66-338 nmol/L; 10-15 years=67-189; adults=67-140 nmol/L.
CSF 3-O-MD: 0-0.2 years≤300 nmol/L; 0.2-0.5 years≤300 nmol/L; 0.5-2 years≤300 nmol/L; 2-5 years≤150 nmol/L; 5-10 years≤100 nmol/L; 10-15 years≤100 nmol/L; adults≤100 nmol/L.
CSF tetrahydrobiopterin: 0-0.2 years=40-105 nmol/L; 0.2-0.5 years=23-98 nmol/L; 0.5-2 years=18-58 nmol/L; 5-10 years=9-40 nmol/L; 10-15 years=9-32 nmol/L; adults=10-30 nmol/L.
CSF neopterin: 0-0.2 years=7-65 nmol/L; 0.2-0.5 years=7-65 nmol/L; 0.5-2 years=7-65 nmol/L; 2-5 years=7-65 nmol/L; 5-10 years=7-40 nmol/L; 10-15 years=8-33 nmol/L; adults=8-28 nmol/L.
Two patients had SSADH deficiency, which was identified by the presence of elevated 4-hydroxybutyric acid in urine organic acid analysis. Direct sequencing of ALDH5A1 (a known pathogenic variantReference Akaboshi, Hogema and Novelletto 6 ) confirmed the diagnosis of SSADH deficiency in both patients. In patient 11, quantification of 4-hydroxybutyric acid in urine was markedly elevated (119 mmol/mol creatinine; reference range 0.09-5). His SSADH activity was non-detectable (reference range 1907-3901 pmol/minute/mg protein) in the cultured lymphoblasts. In additional repeat urine organic acid screens, one demonstrated elevated 4-hydroxybutyric acid and the other was normal.
Treatment and treatment outcomes are listed in Table 1b. Neuropsychological assessment results of patients older than 3 years of age are summarized in Table 1c. Three patients with PTPS, two patients with DHPR and one patient with TH deficiencies had age-appropriate developmental outcome. Because of late diagnosis and/or compliance problems with l-dopa/carbidopa and 5-hydroxytryptophan, one patient with DHPR and one patient with PTPS deficiencies did not achieve normal neurodevelopmental outcome (Table 1c). In one patient with TH and in one patient with AADC deficiencies, medical management resulted in improvements of movement disorders but did not improve neurodevelopmental outcome. All patients with PTPS, DHPR and TH deficiencies were monitored by prolactin levels and maintained normal prolactin levels during outpatient follow-up clinic visits. All patients with PTPS deficiency were on tetrahydrobiopterin once or twice a day and monitored by plasma and blood dot spot phenylalanine target level <360 µmol/L. All patients with DHPR deficiency were on the phenylalanine-restricted diet and achieved plasma phenylalanine target level <360 µmol/L throughout the therapy.
Table 1b Treatment and treatment outcome of patients with inherited neurotransmitter disorders are listed
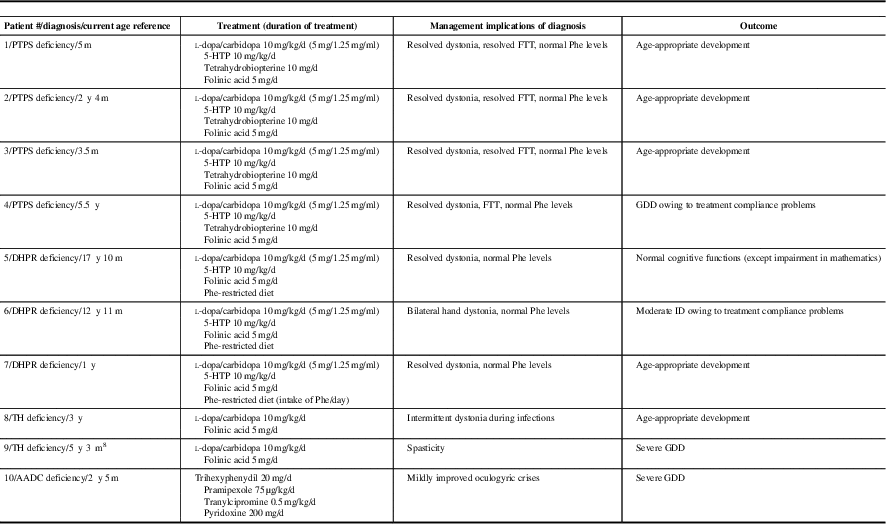
AADC=aromatic l-amino acid decarboxylase; d=day(s); DHPR=dihydropteridine reductase; FTT=failure to thrive; GDD=global developmental delay; 5-HTP=5-hydroxytryptophan; ID=intellectual disability; m=month(s); PTPS=6-pyruvoyltetrahydropterin synthase; TH=tyrosine hydroxylase; y=year(s).
Table 1c Neuropsychological assessment results of patients with inherited neurotransmitter disorders are summarized
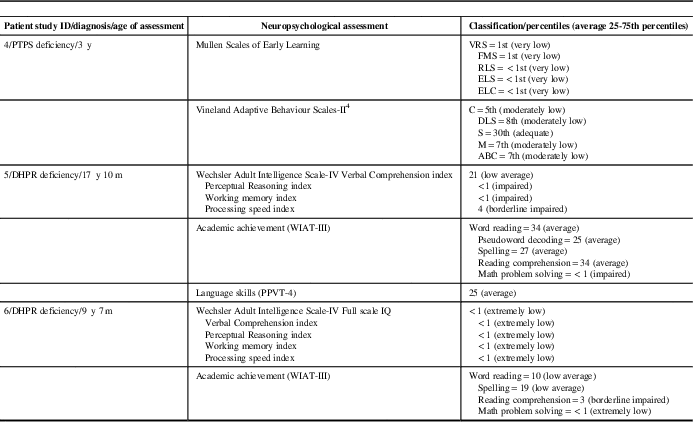
ABC=adaptive behavior composite; C=communication; DHPR=dihydropteridine reductase; DLS=daily living skills; ELS=expressive language score; ELC=early learning composite; FMS=fine motor score; m=month(s); M=motor skills; PPVT-4=Peabody Picture Vocabulary Test-Fourth Edition; PTPS=6-pyruvoyltetrahydropterin synthase; RLS=receptive language score; S=socialization; VRS=visual reception score; WIAT-III=Wechsler Individual Achievement Test-Third Edition; y=year(s).
Mullen Scales of Early Learning normal scores=average 25-74th percentiles. ELC is the total score of all visual, fine motor, receptive language and expressive language. All ELC scores are calculated without the gross motor score.
Vineland Adaptive Behaviour Scales-Second Edition normal scores=average 25-74th percentiles.
Wechsler Adult Intelligence Scale-IV normal scores=mean 100±15 SD.
Wechsler Individual Achievement Test-Third Edition normal scores=Mean 100±15 SD.
The PTPS deficiency was the most common inherited neurotransmitter disorder in our study. The homozygous p.Gly315Ser variant in TH in two unrelated families and the homozygous p.Thr67Met variant in PTS in two cousins from non-consanguineous parents were reported in patients from Sri Lankan ethnic background. These variants are likely to be common founder variants in this population. We report normal neurodevelopmental and neurocognitive outcome in a patient with DHPR deficiency with excellent treatment compliance for 18 years. So far, less than ten patients with DHPR deficiency have been reported for their detailed treatment outcome. Our study reports two additional DHPR patients’ long-term treatment outcome and normal neurocognitive functions in one of those patients. Good treatment compliance is the key for normal neurodevelopmental outcome.
Sixty-eight patients (sum of patients with DHPR and PTPS deficiencies) (12%) were treated from the neonatal period reported in The International Database of Tetrahydrobiopterin Deficiencies.Reference Opladen, Hoffmann and Blau 3 Developmental delay was present in 28%, seizures in 5%, dystonia in 5%, other movement disorders in 20% and autonomic dysfunction in 10% of those patients. In 624 patients, treatment was initiated after the neonatal period. In the latter group, developmental delay was present in 48%, seizures in 28% and autonomic dysfunction in 15% of those patients. The percentage of patients with dystonia and other movement disorders were similar to the patients in whom treatment was initiated in the neonatal period.Reference Opladen, Hoffmann and Blau 3 More than 50% of the patients, treated from the neonatal period, had no symptoms. However, 75% of the patients, treated after the neonatal period, were symptomatic.Reference Opladen, Hoffmann and Blau 3 In our study, 86% (6/7) of the patients were treated from the newborn period and 83% (5/6) of these patients had normal neurodevelopmental outcome. Tetrahydrobiopterin loading test or biochemical investigations including urine neopterin and DHPR activity in blood dot spot in newborns with positive newborn screening for phenylketonuria are essential to confirm diagnosis and initiate treatment in the neonatal period for PTPS and DHPR deficiencies.
Less than 200 patients with SSADH deficiency have been reported. 4-hydroxybutyric acid in urine organic acid analysis and symmetrical increased signal intensity in globus pallidus are typical biochemical and neuroimaging features of SSADH deficiency.Reference Pearl, Wiwattanadittakul, Roullet and Gibson 2 False negative (e.g., hidden 4-hydroxybutyric acid peak by a large urea peak) or false positive (e.g., urine collection by 4-hydroxybutyric acid containing Coloplast SpeediCath catheters) urine organic acid results have been reported to interfere with the diagnosis of SSADH deficiency.Reference Pearl, Wiwattanadittakul, Roullet and Gibson 2 In one of the early case series, several patients were reported to have normal brain MRI.Reference Gibson, Christensen and Jakobs 7 In our study, one SSADH patient had normal urine organic acid analysis and the second SSADH patient had no basal ganglia changes.
More than 100 patients with AADC deficiency have been reported so far. Consensus guidelines for the diagnosis and treatment of AADC deficiency were published recently.Reference Wassenberg, Molero-Luis and Jeltsch 8 The treatment consists of pyridoxine, dopamine agonists, monoamine oxidase inhibitors and anticholinergic agents for symptomatic treatment. Unfortunately, this treatment does not improve neurodevelopmental outcome.
Less than 100 patients have been reported with TH deficiency. The phenotype ranges from dopa-responsive dystonia to severe progressive infantile-onset encephalopathy and Parkinsonism.Reference Furukawa and Kish 9 Patients with severe phenotype develop dopa-induced dyskinesias.Reference Furukawa and Kish 9 Homozygous or compound heterozygous p.Gly315Ser variant results in variable phenotype and variable response to l-dopa/carbidopa therapy based on our, and previously reported, cases.Reference Zhang, Zhou and Li 10
In summary, we report 12 patients with inherited neurotransmitter disorders affecting monoamine, tetrahydrobiopterin and GABA metabolisms and their favorable neurodevelopmental outcome owing to good treatment compliance and early initiation of therapy in patients with PTPS, DHPR and TH deficiencies. All patients with infantile-onset global developmental delay, truncal hypotonia, movement disorders, as well as autonomic dysfunction, should be investigated by CSF neurotransmitter metabolite measurements to be able to initiate early treatment in inherited neurotransmitter disorders affecting monoamine and tetrahydrobiopterin metabolisms. Neither normal urine organic acid analysis nor normal neuroimaging would exclude the diagnosis of SSADH deficiency in patients with global developmental delay.
Acknowledgments
The authors thank Andreas Schulze, Michal Inbar-Feigenberg, Neal Sondheimer, Anette Feigenbaum and James Dowling for referring their patients with inherited neurotransmitter disorders for their management. The authors thank Stacy Hewson for genetic counseling for the genetic test results. The authors also thank Duncan Westwood for his work for Institutional Research Ethics Board application of this study.
Statement of Authorship
DC and GB performed data entry and data analysis, prepared the first draft of the manuscript and approved the final version. RDC and JR carried out diagnosis and follow-up of patients and approval of the final version. SM-A was involved in design and conceptualization of the study, analysis and interpretation of the data and drafting and finalizing the manuscript for intellectual content.
Conflicts of Interest
All of the authors declare no conflicts of interest or competing interests. There were no financial associations.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Ethical Approval
This study is approved by Institutional Research Ethics Board (Approval#1000057003).



