


Introduction
Tumour microenvironment (TME) refers to the cellular environment in which tumour cells exist. The TME is comprised of blood vessels, various molecules, extracellular matrix (ECM) and different kinds of cells including tumour cells, cancer-associated fibroblasts, tumour-associated macrophages (TAMs) and endothelial cells (Ref. Reference Wang1). Although there are many different types of immune cells infiltrating the tumour, macrophages are the most abundant inflammatory cells in human cancers such as bowel, breast, stomach, bladder and lung cancers (Refs Reference Cassetta and Pollard2, Reference Kalbasi3). Substantial clinical and experimental evidence found a close relationship between high infiltration of TAMs and a ‘bad’ prognosis in colorectal cancer, breast cancer and cervical cancer (Refs Reference Wei4–Reference Geng7). However, other studies demonstrated that TAMs infiltration was closely related to a ‘good’ prognosis in ovarian cancer and colorectal cancer (Refs Reference El-Arabey8, Reference Edin9). This suggests that the correlations between TAMs infiltration and clinicopathologic feature are unequal in ‘different types of TAMs’. For instance, TAMs contain two different subpopulations. The first subset is M1 macrophages, which distinguish tumour cells from normal cells and ultimately kill tumour cells; the second subset is M2 macrophages, which harbour pro-tumour functions (Ref. Reference Shapouri-Moghaddam10). However, this classification method is not particularly new and has been used for many years. Additionally, the traditional activation models we used in vitro cannot explain the extent of heterogeneity seen in vivo because of the complexity of TME. For instance, high islet/stromal ratio of macrophages in non-small cell lung cancer (NSCLC) means longer disease-free survival (DFS) and overall survival (OS) in patients (median DFS and OS were 20.7 versus 5.5 months and unreached versus 34.8 months for high islet/stromal macrophage ratio versus low ratio, respectively) (Refs Reference Feng11, Reference Dai12). Apart from locations, the origins and surface markers (programmed death ligand-1 (PD-L1), tyrosine-protein kinase receptor (TIE2) and chemokine (c-c motif) receptor 2(CCR2)) of TAMs also influence their functions and bring difficulties to clinical therapy.
Although multiple studies pointed out that TAMs promoted the recovery of tumours from clinical therapies by promoting angiogenesis and inhibiting immune response, the underlying mechanism remains to be uncovered until now. In some tumours, chemotherapy resistance is always accompanied by accumulated ‘non-sensitive TAMs’ including CD45+CD11b+CD14+ macrophages, MRC1+TIE2HiCXCR4Hi macrophages and CD11b+Ly6C+TIE2+ macrophages. When targeting these accumulated TAMs after chemotherapy, the tumour regrowth and metastasis are significantly decreased (Refs Reference Chen13–Reference Hughes15). Equally, studies in radiotherapy resistance and immunotherapy resistance confirmed the same result. Together, these findings illustrate different subsets of macrophages in regulating therapy responses and unearth that targeting ‘non-sensitive TAMs’ is a novel therapeutic strategy to block resistance. Thus, there is an urgent need to study TAMs heterogeneity and search for novel therapies targeting different subpopulations of TAMs.
Origin and heterogeneity of TAMs
Origin of TAMs
TAMs have two distinct origins: tissue-resident macrophages (TRMs) and bone-marrow-derived macrophages (BMDMs). TRMs, including Kupffer cells, alveolar macrophages and osteoclasts, originate from erythromyeloid progenitors (EMPs) in the yolk sac (C-kit+CD41+EMPs) or foetal liver (colony-stimulating factor 1 receptor-positive EMPs (CSF-1R+ EMPs)). After being stimulated by chemokines in tumours, TRMs accumulate in the target organs and mediate tumour immunity via proliferation. Before the establishment of the blood circulation, EMPs expressing C-kit and CD41 emerge in the yolk sac. These early EMPs rapidly give rise to CSF-1R+ EMPs which could seed in the brain without monocytic intermediates. The CSF-1R+ EMPs can also seed in the foetal liver and then give rise to foetal monocytes. Foetal monocytes then spread to all tissues after the establishment of blood circulation. It is worth mentioning that TRMs cannot seed in the brain after the establishment of the blood-brain barrier (Refs Reference Gomez Perdiguero16, Reference Stremmel17). After birth, haematopoietic stem cells in the bone marrow develop into common myeloid cells and then differentiate into two different monocyte subsets, Ly-6C+ and Ly-6C−. Then, monocytes leave the bone marrow and enter the blood. After being stimulated by chemokines released by the tumour, Ly6C+CCR2+ monocytes traffic into the tumour via the CCL2/CCR2 axis and become mature macrophages named BMDMs (Refs Reference Kalbasi3, Reference Kumar18, Reference Mondini19) (Fig. 1).
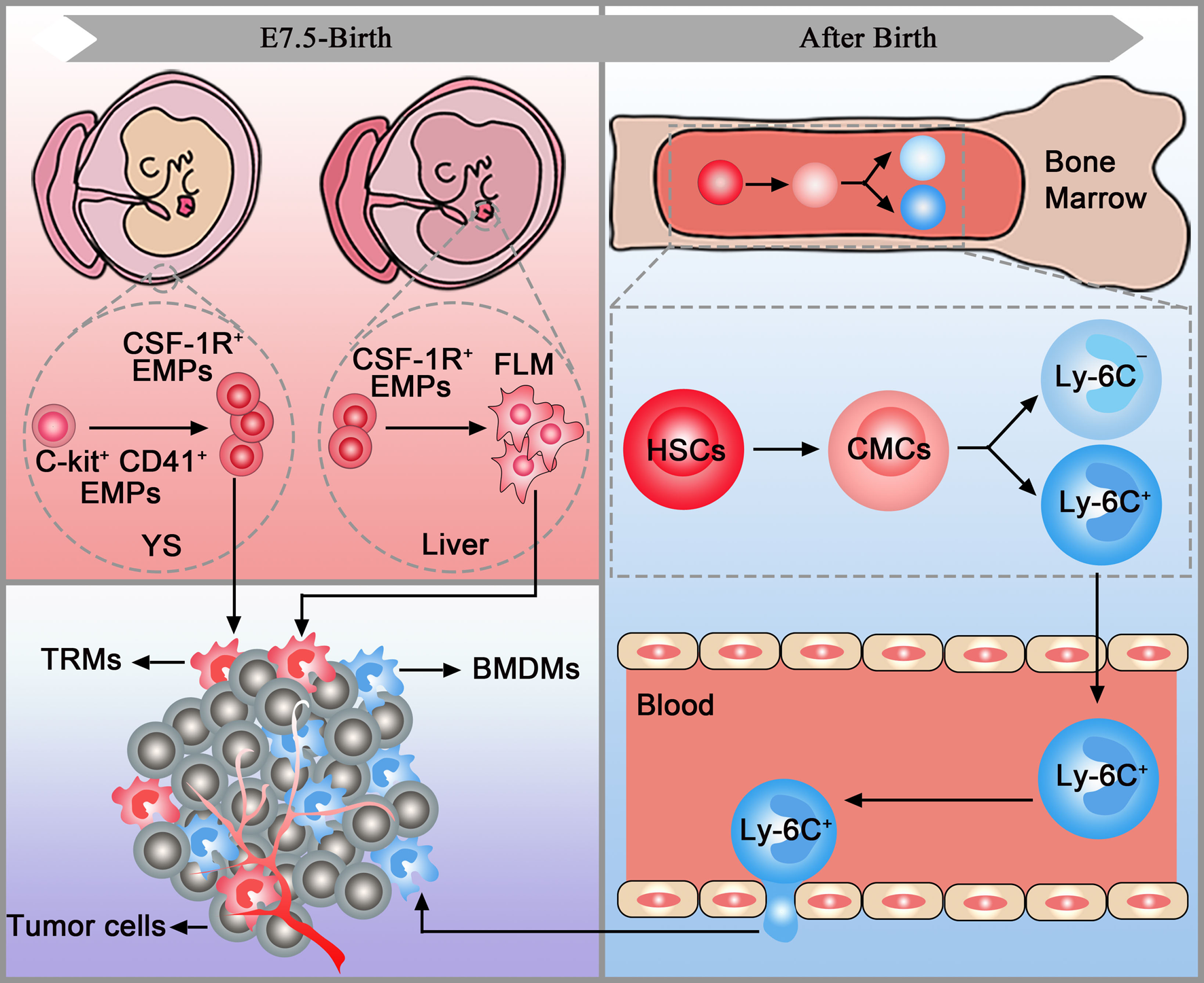
Fig. 1. TAM origins. Tissue-resident macrophages (pink box) and bone marrow-derived macrophages (blue box) are accumulated in the tumour (purple box). YC, yolk sac; FLM, foetal liver monocytes; EMPs, erythromyeloid progenitors; HSCs, haematopoietic stem cells; CMCs, common myeloid cells; TRMs, tissue-resident macrophages; BMDMs, bone marrow-derived macrophages.
In tumours, CD45high macrophages were considered BMDMs, and CD45low cells were considered microglia previously. However, a study in brain malignancies pointed out that CD45 only can be employed in murine models and cannot accurately discriminate microglia and BMDMs in patient samples. Thus, extensive flow cytometry panels are needed to distinguish TRMs and BMDMs in both species. Additionally, CX3C chemokine receptor 1 (CX3CR1), commonly used to trace microglia, cannot be used to discriminate microglia and BMDMs in brain tumours as it is also upregulated in BMDMs (Ref. Reference Bowman20). Similarly, TRMs and BMDMs express comparable levels of CX3CR1 in murine pancreatic ductal adenocarcinoma (PDAC) models (Ref. Reference Zhu21). Further studies found that multiple markers, including CXCR4, Itga4 (CD49D), CD11b and CD11a, can be used to distinguish TRMs and BMDMs in tumours (Refs Reference Bowman20, Reference Zhu21).
Phenotypic heterogeneity of TAMs
After being stimulated by chemokines in the TME, these extremely plastic macrophages polarise into M1 and M2 subtypes (Ref. Reference Komohara22). Lipopolysaccharide (LPS), granulocyte-macrophage colony-stimulating factor, interferon γ (IFN-γ), and activated Toll-like receptors (TLRs) stimulate macrophages to M1 subpopulations. M1 macrophages are considered as anti-tumour macrophages because they contribute to innate host defense and killing tumour cells by producing reactive oxygen/nitrogen species and pro-inflammatory cytokines (e.g., interleukin-1β (IL-1β), IL-6, IL23 and tumour necrosis factor α (TNF-α)) (Ref. Reference Jeannin23). To identify the M1 subtype from macrophages, markers for the M1 subtype, such as CD80/CD86, iNOS, HLA-DR and pSTAT1, were used in recent studies. On the contrary, Th2 cytokines such as IL-4, IL-10 and IL-13 promoted the macrophages to M2 polarisation, which was considered pro-tumour macrophage because they produced cytokines such as IL-10, IL-13 and transforming growth factor β (TGF-β) to promote tumour development. Similarly, markers for M2 TAMs, such as arginase 1 (Arg1), CD206, CD204 and CD163, were used to identify M2 subtypes. M2 macrophages can be further subdivided into M2a, M2b, M2c and M2d subpopulations depending on the function and cell surface markers they display in vitro. M2a macrophages are stimulated by IL-4 and IL-13. In addition to expressing high levels of the decoy IL-1 receptor (IL-1R), mannose receptor (MR or CD206) and CCL17, M2a macrophages also secrete insulin-like growth factor (IGF), fibronectin and TGF-β. Another subpopulation, M2b macrophages are stimulated by IL-1R agonists or TLR ligands and express high levels of TNF superfamily member 14 (TNFSF14) and CCL1. After being stimulated by IL-10, glucocorticoids or TGF-β, M2c macrophages secrete a high level of IL-10 and then participate in anti-inflammatory activities. M2d macrophages, closely interacted with tumour angiogenesis and progression, are induced by TLR agonists (Ref. Reference Colin, Chinetti-Gbaguidi and Staels24). However, little progress has been made on the characterisation of macrophages in vivo. In vitro assay allows easy control of the extracellular environment and can be used for the analysis of macrophage polarisation. Conversely, in vivo conditions contain a variety of cytokines secreted by other immune cells and could affect the outcome of macrophage polarisation, making classification difficult. Until now, TAMs only can be classified into M1 macrophages and M2 macrophages in vivo. Recently, Nikovics et al. identified M2d-like macrophages in the minipig model by in situ hybridisation, providing more possibility that the M2 classification system can be also used in vivo (Ref. Reference Nikovics25).
Both M1 and M2 macrophages are highly plastic and thus can evolve into each other after TME changes or therapeutic interventions. In the early stage of the tumour, M1 TAMs account for a relatively high proportion, while the proportion of M2 TAMs is higher in advanced tumours (Ref. Reference Singhal26). During tumour progression, monocytes secrete CXCL12 into the TME and then promote the transition from M1 to M2 macrophages (Ref. Reference Sánchez-Martín27). In contrast, blocking polarisation factors, such as PI3K-γ pathway, CD40, STAT3, STAT6, CSF-1/CSF-1R, result in a switch from M2 to M1 macrophages, providing a target for clinical therapy (Refs Reference Li28–Reference Poh and Ernst30). Recently, a group achieved TAMs-specific delivery and M2 to M1 transition through liposome, providing more possibility for future therapy (Ref. Reference Li, Somiya and Kuroda31). Interestingly, TAMs from murine fibrosarcoma not only express a high level of M2 phenotype, but also express proinflammatory Th1 chemokines like CCL5, CXCL9 and CXCL10, indicating the presence of a ‘mixed’ macrophage phenotype in tumours (Ref. Reference Biswas32). As expected, single-cell RNA sequencing of NSCLC did not find any macrophages only exhibiting the M1 phenotype (Ref. Reference Zilionis33). Therefore, simply classifying macrophages into M1 and M2 cannot cover all types of TAMs.
Apart from the conventional binary model, which classified macrophages into M1 and M2 subtypes, a single cell atlas in breast cancer demonstrated that the heterogeneously PD-L1+ TAMs can be classified into seven different clusters: M01 (CD38, CD163, CD169, CD204, CD206), M02(CD169, CD204), M04, M14, M17(CD38, CD169), CD64highHLA-DRhigh. This classification model is closely related to clinical therapy as ER− (oestrogen receptor-negative) tumours contained more M01 and M17 TAMs than ER+ tumours. In addition, grade 3 tumours contain fewer M04 TAMs than lower-grade tumours (Ref. Reference Wagner34). In addition, Arlauckas et al. proved the existence of elongated Arg1A+ macrophages and rounded Arg1B+ macrophages in the tumour and found that these macrophages were located in the tumour periphery and tumour centre respectively (Ref. Reference Arlauckas35). Among the human TAMs subset in NSCLC, they divided TAMs into 10 different groups expressing distinct chemokines (G1 expressing CXCL5; G8 expressing CXCL12; G9 expressing CXCL9, CXCL10 and CXCL11) (Ref. Reference Zilionis33). With different TAMs subsets and labelling markers defined here, we can test the correlation of different subgroups to tumour progression and provide new targets for clinical therapy in the future.
Diversity of TAMs function
According to the previous studies, we can classify TAMs into the following six types: anti-tumour macrophages, immunosuppressive macrophages, invasive macrophages, metastasis-associated macrophages, angiogenic macrophages and lymphangiogenic macrophages (Ref. Reference Qian and Pollard36).
M1-macrophages, also named anti-tumour macrophages, can identify tumour cells from normal cells and ultimately kill tumour cells through two different mechanisms. In the first mechanism, M1-macrophages secrete tumour-killing molecules such as ROS and NO, killing tumour cells directly through cytotoxic effects (Ref. Reference Bernsmeier, van der Merwe and Périanin37). The other mechanism is antibody-dependent cell-mediated cytotoxicity killing tumour cells. Recently, a study pointed out that M1-derived exosomes exerted a proinflammatory role and suppressed angiogenesis in myocardial infarction, revealing that M1 macrophages may inhibit tumours by molecules carried by exosomes (Ref. Reference Liu38). Further studies proved that M1-derived exosomes act as the carrier to deliver paclitaxel (PTX) and cisplatin (DDP) into the tumour tissues, enhancing the anti-tumour effects in the mice model (Refs Reference Li, Li and Wang39, Reference Wang40). Thus, M1-derived exosomes may be an effective approach to treat cancer cells in the future. Interestingly, increasing evidence revealed that M1 macrophages also promoted tumour development. During tumour initiation, TAMs create an inflammatory environment that is mutagenic and promotes growth. In 1863, Rudolf Virchow noted leucocytes in neoplastic tissues and made a connection between chronic inflammation and cancer. Over the past few decades, multiple studies demonstrated that cancer is associated with infection (viral hepatitis B, viral hepatitis C, Helicobacter pylori) and inflammation is widely involved in the process of carcinogenesis as the primary mediator (Ref. Reference Quante and Wang41). As a matter of course, infection treatment (i.e., H. pylori in gastric cancer) and various anti-inflammatory therapies (i.e., COX-2 inhibitors) can successfully prevent cancer progression (Refs Reference Marcelis, Tousseyn and Sagaert42, Reference Mahboubi Rabbani and Zarghi43). During chronic inflammation, a mutagenic and growth-promoting environment was created in the subepithelial stroma, which was infiltrated by various inflammatory cells and activated macrophages account for the majority of the immune cells. Then, these activated macrophages generated ROS and NO intermediates which induced DNA damage in proliferating cells and the surrounding epithelial cells, supporting tumour initiation and neoplastic transformation (Ref. Reference Balkwill, Charles and Mantovani44). Moreover, M1 macrophages potently induced epithelial-mesenchymal-transition (EMT) in benign and malignant PDAC cell lines (Ref. Reference Helm45). Besides, a recent study showed that supernatant from the M1 macrophage culture contained cytokine IL-1β which controls PD-L1 expression in hepatocellular carcinoma cells (Ref. Reference Zong46). The macrophages used in these studies were cell lines or murine BMDMs which were different from M1 TAMs. Without considering this defect, we speculate that there exist ‘mixed’ TAMs in tumours because polarisation is driven by various environmental factors.
TAMs downregulate the activation of other immune cells and suppress the immune response by secreting various cytokines (vascular endothelial growth factor (VEGF), IL-4, IL-10 and TGF-β), chemokines (CCL2, CCL18 and CCL22) and enzymes (COX-2, ARG1 and matrix metalloproteases (MMPs)). TAMs suppress immune response through two different mechanisms: (1) inhibiting the function of CD8+ and CD4+ T cells; (2) promoting the function of the induced regulatory T cells (i-Tregs) and natural Tregs cells (n-Tregs). For instance, TAMs-derived IL10 and TGF-β promoted the production of i-Treg in the intestinal (Ref. Reference Denning47). In turn, Tregs in tumours promoted the polarisation of M2 macrophages (Ref. Reference Liu48). M2 macrophages expressed proteins, such as epithelial growth ligands of the factor, CSF-1, cat eye syndrome critical region protein 1, are shown to stimulate the migration of glioma cells (Refs Reference Zhu49–Reference Yin51). Macrophage-derived exosomes, which were considered to be a key factor in mediating the communication between tumour cells and microenvironment cells, can also promote the migration of tumour cells as it carries multiple microRNAs and proteins, such as miR-21-5p, miR-155-5p and ApoE protein (Refs Reference Lan52, Reference Zheng53). Besides, TAMs possess the ability to promote EMT by activating the COX-2/STAT3 axis (Ref. Reference Han54). During EMT, the interaction between cell-cell and cell-ECM are reshaped, resulting in the separation of epithelial cells from each other and the underlying basement membrane, in other words, increasing the migration capacity of tumour cells (Ref. Reference Dongre and Weinberg55). Apart from moving tumour cells, a destroyed basement membrane is a requirement for tumour metastasis. M2 macrophages can destroy endothelial cell basement membrane by secreting matrix serine proteases, MMPs, cathepsins, etc., and decompose various collagen and other components of ECM, thereby increasing the migration of tumour cells (Refs Reference Hao56, Reference Annamalai57).
The formation of new blood vessels is essential to provide nutrients and oxygen to malignant cells. In animal models of prostate cancer, breast cancer and cervical cancer, TAMs promoted tumour angiogenesis in a VEGF-dependent manner (Refs Reference Halin58–Reference Zhang61). The expression of the two transcription factors, hypoxia-inducible factor-1α (HIF-1α) and HIF-2α, within hypoxia TAMs is the key to VEGF production, and knockout of these proteins in TAMs attenuates pro-angiogenic responses (Refs Reference Werno62, Reference Niu63). Further study revealed that HIF-1α induced recruitment of CD45+ myeloid cells containing TIE2+ subgroups (Ref. Reference Du64), and knockdown of TIE2-expressing macrophages in mice suppressed angiogenesis by reducing VEGF and autophagy, indicating a HIF-1α/TIE2/VEGF pathway in angiogenesis (Ref. Reference Zhang65). Tumour-released CSF-1 (M-CSF) also regulated VEGF-A expression through NF-κ B and CCL2, promoting pro-angiogenic functions of macrophages (Ref. Reference Wyckoff66). Another cytokine, MMPs, can be released by TAMs and contributes to the release of bioactive VEGF-A from its ECM-bound form (Ref. Reference Riabov67). Then, bioactive VEGF stimulates the vascular permeability of blood vessels and increases the extravasation of plasma proteins, thereby providing a scaffold for the migration of activated endothelial cells. Considering the central role of VEGF in angiogenesis, the combination of docetaxel and ramucirumab, inhibitors of the VEGF/VEGFR2 pathway, is applied for second-line therapy in NSCLC (Refs Reference Yoh68, Reference Villaruz and Socinski69). Although the progression-free survival and OS improved after treatment, the median survival (MS) of the NSCLC patients is shorter than 18 months. These results indicate that VEGF-independent angiogenic factors also promote angiogenesis. Indeed, TAMs release a panel of VEGF-independent angiogenic factors, including semaphorin 4D and urokinase-type plasminogen activator (Refs Reference Zhang70, Reference Binmadi71). Recently, a study about PDAC found that M2 macrophage-derived exosomes which carry a higher level of miR-155-5p and miR-221-5p than M0 macrophages-derived exosomes increased the vascular density of mice (Ref. Reference Yang72).
Lymphangiogenesis is essential for tumour cells lymph node metastasis. TAMs can utilise two main mechanisms to stimulate lymphangiogenesis. Firstly, malleable TAMs promote lymphangiogenesis by trans-differentiation into lymphatic endothelial cells. It was reported that LYVE-1+TAM integrated into tumour-associated lymphatic endothelium in mouse models of pancreatic insulinoma and prostate cancer (Ref. Reference Zumsteg73). Secondly, TAMs directly secrete pro-lymphangiogenic factors. For instance, M2 TAMs can induce the development of lymphatic vessels in the tumour by secreting pro-angiogenesis factors, such as VEGF and MMP9 (Ref. Reference Fu74). Podoplanin positive (PDPN+) TAMs also promoted lymphangiogenesis and lymphoinvasion in cancer (Ref. Reference Bieniasz-Krzywiec75). Recently, a study pointed out that TRMs were closely related to lymphatic vessel growth and expansion in the heart. Even without using tumour models, this finding reveals the relationship between a subpopulation of macrophages and lymphangiogenesis and provides insight into our future study (Ref. Reference Cahill76).
Cause of TAMs heterogeneity
Multiple tumours
What are the factors that cause the heterogeneity of TAMs? Firstly, the functional heterogeneity could be determined by the sources of TAMs. For example, Loyher et al. demonstrated that TRMs were associated with tumour cell growth in lung cancer, while CCR2-dependent recruited macrophages accelerated tumour progression in the form of metastasis (Ref. Reference Loyher77). In murine PDAC models, a recent study showed that embryonically derived TAMs exhibited pro-tumoural function whereas their bone marrow macrophage counterparts mainly contributed to antigen presentation, suggesting the importance of origins (Ref. Reference Zhu21). Moreover, CD163+ Tim4+ macrophages in the omentum which originated from embryos promote the development and metastasis of ovarian cancer (Ref. Reference Etzerodt78). Moreover, the proportion of TRMs and BMDMs in tumours varies in different tissues and organs respectively. Franklin et al. found that CCR2+ monocytes accounted for approximately 40% of all CD45+ cells whereas only 10% of cells derived from TRMs in breast cancer (Ref. Reference Franklin79). Conversely, TAMs are mostly derived from TRMs in retinoblastoma, neuroblastoma and osteosarcoma (Ref. Reference Ostuni80). These observations provoke a question: whether TAMs from different sources, such as TRMs (generally express CD11b and MERTK) and BMDMs can be individually targeted (Refs Reference Bowman20, Reference Gautier81)? As expected, MERTK inhibition combined with radiotherapy showed a significant growth delay than radiotherapy alone in mouse glioblastoma which mainly contains TRMs (Ref. Reference Wu82). Specific depletion of CD163+ Tim4+ macrophages also prevents metastasis of ovarian cancer. In addition, inhibition of BMDMs delays the glioblastoma recurrence after radiotherapy because the abundance of BMDMs increased after radiotherapy (Ref. Reference Akkari83). However, some BMDMs probably convert into TRMs during growth, meaning the conversion of functions and also bring difficulties for targeted therapy based on TAM origins (Ref. Reference Moore, Sheedy and Fisher84).
Secondly, macrophage subpopulations are not uniformly distributed along the tumour. In stage II colorectal tumours, macrophages carrying CD163 are predominantly located in the tumour invasive front (IF) whereas macrophages carrying CD80 are mainly found within the tumour-adjacent normal mucosa (Ref. Reference Pinto85). Another study in colorectal tumours found a higher level of CD163+/CD68+ ratio in tumour IF than that in tumour centre. Additionally, a higher level of CD163+/CD68+ ratio in tumour IF was closely associated with enhanced LVI, TI and TNM stage (Ref. Reference Yang86). In breast cancer, the distribution of CD86+ and CD163+ within tumour nest (TN) and tumour stroma (TS) is also different. In the same study, they revealed that high density of CD163+ TAM within TS was closely correlated with poor prognostic parameters, whereas there was no association with CD163+ TAMs density and clinicopathologic characteristics in TN (Ref. Reference Mwafy and El-Guindy87).
Third, the distribution of macrophages varies in different tumours. In breast tumours, the high infiltration of CD68+ TAMs in TN and TS is correlated with large tumours. However, another study in colorectal cancer found that low infiltration of CD68+ TAMs in IF and TS was associated with more advanced cancer (Refs Reference Gulubova88, Reference Zhou89). Considering that the CD68+ marker is used to define the total amount of TAMs, the CD68+ TAMs might represent different subpopulations in different tumours. Additionally, distinct structures and molecular compositions between tissues might regulate local immunity and influence the antitumour immune response across tumour sites (Ref. Reference Salmon90). For example, the densities of the lymphoid structures (TLSs) which differ between tumours reflect lymphocyte-infiltration levels (Ref. Reference Buisseret91).
Multiple stimulations of bioactive molecules
LPS/IFN-γ polarisation pathway
IFN-γ and LPS are classical stimuli for M1 macrophages. During M1 polarisation, IFN-γ and LPS can bind to receptors at the membrane and subsequently activate the downstream molecules. TLRs regulate the polarisation of macrophages through MyD88-dependent and MyD88-independent pathways. In the MyD88-dependent pathway, TLRs signal activates the NF-κB through the adaptor protein MyD88 and κB kinase (IKK) complex which consists of IKKα and IKKβ, and IKKγ (or NEMO). As a result, NF-kB (p65/p50 complex) can be released from cytoplasmic NF-kB/I-kB complex and rapidly translocate in nuclear and bind to target genes such as TNFα and IL-1. Moreover, MyD88 also activates the transcription factor, AP-1, by triggering the activation of MAPK (Fig. 2a). In the MyD88-independent pathway, macrophages polarisation is regulated via TLRs-TRIF-IKKi/TBK1-IRF3 pathway. After being stimulated by TLRs, TIR-domain-containing adapter inducing interferon-β (TRIF) induces the activation of IKKi and TBK1 and then regulates the interferon-regulatory factor 3 (IRF3) (Fig. 2b). Another polarisation factor, IFN-γ, binds to its cognate receptor (IFNAR1/2) and regulates macrophages polarisation through the IFNAR1/2-Jak-STAT1 pathway (Ref. Reference Hagemann92) (Fig. 2c). The Notch pathway is also involved in M1 polarisation. In 2010, Wang et al. provided new insight into the molecular mechanism of macrophages polarisation and found that compromised Notch pathway activation led to the M2-like TAMs (Ref. Reference Wang93). Research in the past 10 years demonstrated that Notch activation promotes M1 polarisation through the Notch1-NF-κB axis pathway (Refs Reference Lin94, Reference Zhou95).
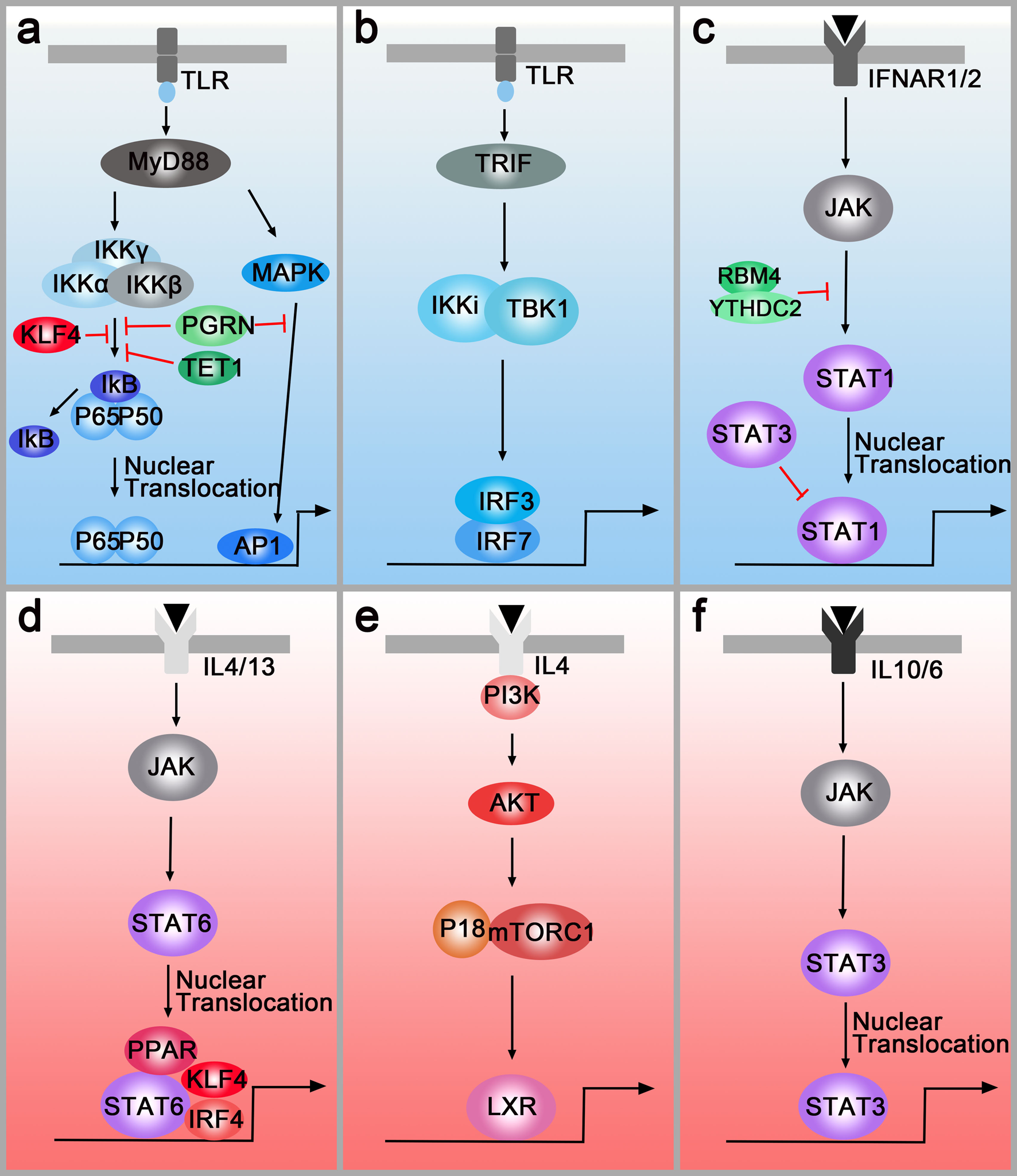
Fig. 2. M1 polarisation pathways (blue box) and M2 polarisation pathways (pink box). (a) Left panel: TLR-MyD88-IKK-NF-κB pathway. Right panel: TLR-MyD88-MAPK-AP1 pathway. PGRN, TET1, RBM4 inhibit M1 polarisation by down-regulating the MyD88-dependent pathway. (b) TLRs-TRIF-IKKi/TBK1-IRF3/IRF7 pathway. (c) IFNAR1/2-Jak-STAT1 pathway. RBM4/YTHDC2 and STAT3 regulate M1 polarisation by targeting the STAT1. (d) IL4/IL13-Jak-Stat6 pathway. PPAR, IRF4 and KLF4 can cooperate with STAT6 and activate M2-type genes. (e) IL4-AKT-P18/mTORC1-LXR pathway. (f) IL-6/IL10-JAK-STAT3 pathway.
These pathways can be regulated by multiple molecules. For example, PGRN and TET1 inhibit M1 polarisation by down-regulating the MyD88-dependent pathway (Refs Reference Liu96, Reference Huang97) (Fig. 2a), and RNA-binding motif 4 regulates M1 polarisation through targeting STAT1 (Ref. Reference Huangfu98) (Fig. 2c). Additionally, multiple studies indicated that epigenetic regulation factors also modulated macrophage polarisation. For instance, N 6-methyladenosine-forming enzyme METTL3 regulates STAT1 mRNA and protein expression through methylation, changing macrophages polarisation (Ref. Reference Liu99). Other methylation enzymes, DNA-methyltransferase-3β and histone deacetylase-3, can also regulate macrophages polarisation (Refs Reference Wang100, Reference Mullican101).
IL4/IL13/IL10 polarisation pathway
IL-4 and IL-13 are overexpressed in human and murine cancer and have been sown to play critical roles in modulating the immune system for tumour growth. Similarly, IL-4R and IL-13R are also highly expressed in various tumours. IL-4R and IL-13R share two chains and initiate signal transduction (Ref. Reference Junttila102). In the first chain, IL-4 binds to a complex composed of IL-4Rα and common cytokine receptor γ-chain. In the second chain, IL-4Rα and IL-13Rα1 form a receptor complex that can bind to both IL4 and IL13 (Ref. Reference Junttila102). Then, the receptors regulate the polarisation of macrophages through two different pathways. In the first pathway (IL4/IL13-Jak-STAT6 pathway), the receptors activate the transcription factor STAT6, resulting in the translocation of pSTAT6 to the nuclei (Ref. Reference Binnemars-Postma103). Besides, the peroxisome proliferator-activated receptor, IRF4 and Krüppel-like factor 4 (KLF4) can cooperate with STAT6, activating M2-type genes such as Arg1, IL10 and Mrc1 (Refs Reference Gao104–Reference Satoh106) (Fig. 2d). Both in vitro and in vivo assays have demonstrated that inhibition of the JAK/STAT pathway in TAMs is critical to attenuate tumour growth and metastasis (Refs Reference Binnemars-Postma103, Reference Suzuki107). In the second pathway (IL4-AKT-P18-mTOR-LXR pathway), AKT will be activated by PI3K when the macrophages are treated with IL-4. Then, P18 interacts with amino acid-activated mTORC1 and activates LXR which will promote M2 gene expression (Ref. Reference Bi108) (Fig. 2e). On the membrane surfaces, M2 macrophages-derived IL10 binds to the IL10R which recruits cytoplasmic protein JAK to the surface. Then, STAT3 is phosphorylated by JAK and translocates to the nucleus, facilitating transcriptional regulation of target genes such as VEGF-A. Additionally, a novel mechanism of M2 polarisation, the IL-6/STAT3 signalling pathway, is found in hepatocellular carcinoma (Ref. Reference Yin109) (Fig. 2f).
Interestingly, the M1 and M2 polarisation pathways can also be regulated by each other. In microglia, IRF-3 (involved in the M1 polarisation pathway) is shown to activate PI3K/AKT signalling (M2 polarisation pathway), suggesting that IRF-3 promotes switching from monocytes to M2 macrophages (Ref. Reference Tarassishin, Suh and Lee110). In natural killer cells and macrophages, ablating of STAT3 upregulates STAT1 expression and increases anti-tumour immune responses, demonstrating the ability of STAT3 to antagonise STAT1 (Ref. Reference Yu, Pardoll and Jove111). Apart from cooperating with STAT6 to induce M2 polarisation, KLF4 inhibits M1 targets via sequestration of coactivators required for NF-κB activation (Ref. Reference Liao112) (Fig. 2a). However, there is no paper studying the preference of polarisation pathways until now. For this reason, we choose to add the urgent need for studying the preference of polarisation pathways in different tumours.
Transcription regulation of macrophages polarisation by RNAs
Recent studies have also demonstrated that microRNAs, a group of small non-coding RNAs, control macrophage polarisation by binding to the 3′ untranslated region of mRNAs to target them for translational repression or degradation. For example, miR21 and miR155 promote M1 polarisation by regulating the JAK/STAT1 pathway whereas miR222-3p promoted M2 polarisation through the JAK/STAT3 pathway. Some microRNAs, such as miR125 and miR26a, suppress M2 polarisation through directly interacting with transcription factors IRF4 and KLF4, respectively (Ref. Reference Li113). Long non-coding RNA also modulates macrophages polarisation in tumours (Refs Reference Tao114, Reference Cao115). Additionally, exosomes can play an important role in macrophages polarisation when it is carrying these regulation RNAs (Ref. Reference Pritchard116). Although we have a little understanding of the polarisation pathways right now, the organ is complicated and regulated by various factors inside and outside the body and one pathway is not enough to understand macrophages polarisation. Therefore, an in-depth understanding of polarisation pathways and their regulatory relationships is essential for later study. If the central regulator of macrophages polarisation can be found, it will be a huge guiding role in clinical treatment.
Multiple metabolisms
Hypoxia results in the generation of ROS, which subsequently oxidise proteins and cause DNA damage, causing genomic instability and then accelerating malignant progression. In recent years, numerous studies have demonstrated that hypoxia promoted increased M2 macrophage in TME. On the one hand, high concentrations of chemokines secreted from hypoxic tissues such as HIF-1, HIF-2, ZEB1 and endothelin-2 attract macrophages into the tumour (Ref. Reference Murdoch, Muthana and Lewis117). Once macrophages arrived in these hypoxic regions, they accumulate in hypoxia tissues by two plausible mechanisms (a) cytokine gradient attracts macrophages and (b) the mobility of macrophages within these areas is disturbed (Ref. Reference Henze and Mazzone118). On the other hand, tumour hypoxia promotes the polarisation of M2 macrophages. As a byproduct of hypoxic glycolysis, tumour-derived lactic acid has a critical function in M2 macrophages polarisation in a HIF1α-dependent manner (Ref. Reference Colegio119). Oncostatin M, eotaxin and neuropilin-1 have also been shown to promote M2 polarisation (Refs Reference Tripathi120, Reference Chen121). Furthermore, both in vitro and in vivo assay found that hypoxia-induced exosomes have a potent ability to promote M2-like polarisation as it carries immunomodulatory proteins and chemokines including FTH, FTL, CCL2, TGF-β and CSF-1 (Ref. Reference Park122). Macrophages preferentially accumulate in hypoxia tumour areas once they are polarised to M2 which can ‘support’ the tumour in overcoming the hypoxic environment and thus maintain its progression (Ref. Reference Lima123). Interestingly, a recent study demonstrated that hypoxic TAMs were more proximal to the lymphatic vessels than normoxic TAMs, suggesting that these hypoxic TAMs might promote cancer progression and metastasis (Ref. Reference Chen124). Although MHC-IIlo M2 and MHC-IIhi M1 are located in strong hypoxia and less hypoxic areas, respectively, hypoxia does not affect macrophages' differentiation and polarisation as the relative abundance and expression markers of TAMs did not change in the hypoxia changing area (Ref. Reference Laoui125). Thus, the phenotypic switches in TAMs are dependent on cytokine availability in conjunction with microenvironmental cues such as hypoxia which is a common feature in many pathophysiological settings. In mice subcutaneous tumours and human NSCLC, on the opposite, TAMs enhance tumour aerobic glycolysis and hypoxia, complicating tumour response to anticancer therapies (Ref. Reference Jeong126).
Another factor, iron homeostasis, also influences macrophages polarisation. In RAW264.7 cells, they discovered high levels of iron storage (ferritin) in M1 macrophages (Ref. Reference Gan127). When exploring the relevance of iron homeostasis and macrophages polarisation in patients, a study in 102 human NSCLC found a survival advantage in patients with accumulating iron compared to iron-negative patients (Ref. Reference Thielmann128). Additionally, iron-positive tumour tissues showed a higher infiltration of M1 TAMs which may contribute to the positive effect on the OS of NSCLC patients. Similarly, a study found that hepatocellular carcinoma cells upregulated the expression of M2-related genes by competing with macrophages for iron (Ref. Reference Sun129). Then, declined iron promoted M2 TAMs polarisation in a prolyl hydroxylase domain (PHD)-dependent manner. Ferrous iron is essential for PHD hydroxylase activity which controls HIF-1α degradation in a post-transcriptional manner. Thus, iron chelated melanin-like nanoparticles limited malignant metastasis and tumour growth by enhancing M2-to-M1 repolarisation (Ref. Reference Rong130). However, a study in cell lines illustrated that chronic iron overload promoted M2 polarisation. And a meta-analysis revealed a positive relationship between haeme iron intake and breast cancer risk (Ref. Reference Chang, Cotterchio and Khoo131). Here, we conclude two possible explanations for these converse results. On the one hand, macrophages, in cell lines, take iron via endocytosis, but no haeme metabolism, whereas macrophages in vivo predominantly take up iron by haeme metabolism. On the other hand, there is a balance between iron homeostasis and tumours. Both lacking and overload of iron are negative to patients.
Recently, a study about manganese (Mn2+) found that Mn-deprived mice had significantly promoted tumour growth and metastasis and Mn2+ treatment significantly reduced tumour burdens in mice (Ref. Reference Lv132). As expected, Mn2+ treatment promotes M1 TAM polarisation and promotes macrophages to produce huge amounts of type I IFNs (Ref. Reference Song133). More importantly, a combination treatment of anti-PD-1 antibody and Mn2+ in patients with advanced metastatic solid tumours shows promising consequents. This work broadens the repertoire of anti-cancer therapy and guides exploring the relationship between Mn2+ and macrophages.
Consequences of TAMs heterogeneity
Chemotherapy resistance
Reciprocal interactions between cancer cells and immune cells in the TME not only support tumour development and progression but can also contribute to resistance to anticancer targeted therapies (Ref. Reference Christie and Bowtell134). Initially in mouse models, CSF1 inhibition was able to reverse chemoresistance (CMF: cyclophosphamide, methotrexate, 5-fluorouracil; cycled twice i.p.) of breast cancer (Ref. Reference Paulus135). Subsequently, DeNardo et al. found a higher percentage of infiltrated CD45+CD11b+CD14+ macrophages in breast cancer biopsy samples from patients who were treated with neoadjuvant chemotherapy than those who received surgery alone. In the same study, mammary epithelial cells produced monocyte/macrophage recruitment factors, including CSF1 and IL-34, after chemotherapy. Blockade of macropage recruiement with anti-CSF1R antibodies, in combination with paclitaxel, improved chemosensitivity and resulted in reduced primary tumour development (Ref. Reference DeNardo14). Additionally, MRC1+ TIE2Hi CXCR4Hi macrophages are accumulated around blood vessels in breast tumours after chemotherapy (Doxorubicin), and these accumulated macrophages are thought to promote tumour revascularisation and relapse via the VEGF-A pathway. When targeting this accumulated TAMs population around blood vessels by blocking CXCR4 after chemotherapy, the tumour revascularisation and regrowth were significantly decreased, suggesting that this targeting strategy is of clinical utility (Ref. Reference Hughes15). Another study found abundant CD11b+ Ly6C+ macrophages infiltrated in tumours after chemotherapy. These CD11b+ Ly6C+ macrophages also highly expressed TIE2 which prevented macrophages from apoptosis under stress via the AKT-dependent pathway (Ref. Reference Chen13). Additionally, Shree et al. also found increased macrophage infiltration following paclitaxel (Taxol) chemotherapy in the mammary tumour. They demonstrated that cathepsin secreted by macrophages interacted with cathepsin inhibition in vivo and prevented Taxol-induced tumour cell death (Ref. Reference Shree136). In addition, TAM-derived CCL2 activated the PI3K/AKT/mTOR signalling pathway in breast cancer cells and then increased endocrine resistance (Ref. Reference Li137). Other cytokines, such as IL6, IL10 and IL34, were reported to mediate chemotherapy resistance in many tumours by inducing anti-apoptotic programs (Refs Reference Kong138–Reference Baghdadi140). Recently, a study found that macrophages-derived YKL-39 which stimulated angiogenesis elevated in mammary patients who undergo neoadjuvant chemotherapy. Thus, anti-angiogenic therapy combined with neoadjuvant chemotherapy provides a new orientation for reducing chemoresistance and inhibiting metastasis in breast cancer patients (Ref. Reference Kzhyshkowska, Larionova and Liu141) (Fig. 2 blue box).
Although both TRMs and BMDMs are depleted after chemotherapy, only BMDMs recover rapidly and undergo phagocytosis-mediated tumour clearance. As TAMs have been shown to express VEGFR1, Loyher et al. speculated that anti-VEGF combined with chemotherapy could target recovered BMDMs and improve therapeutic outcomes. As expected, combined therapy inhibits both TRMs and BMDMs reconstitution through blocking TAM's differentiation and/or survival (Ref. Reference Loyher77). Treatment with cisplatin or carboplatin increases the proportion of M2 macrophages through IL-6 and prostaglandin E2 in ovarian cancer cell lines (Ref. Reference Dijkgraaf142). In the end, M2 macrophages, in turn, promote the chemoresistance of cancer cells in positive feedback. At the transcription level, M2 macrophages upregulate the expression of drug resistance genes, ABCG2, in ovarian cancer cell lines (Ref. Reference Mlynska143).
Radiotherapy resistance
Consistent with chemotherapy, radiotherapy will increase the expression of monocyte/macrophage recruitment factors, including CSF1, CCL2 and IL-34 (Refs Reference Kalbasi3, Reference Stafford144). When combining radiotherapy with CSF-1R inhibitors for tumour therapy, the MS of mice was significantly prolonged than radiotherapy alone. In further studies, they found that CSF-1R inhibitors prevented radiotherapy-recruited monocyte cells from differentiating into M2 TAMs and increased M1 phenotype genes, especially IL-6 and nitric oxide synthase 2 (Ref. Reference Stafford144). Similarly, when co-culturing the inflammatory breast cancer with macrophages, the M1 macrophages mediated radiosensitivity of inflammatory breast cancer cells whereas M2 macrophages mediated radiation resistance. To further clarify the underlying cause of M2 macrophages-mediated radiation resistance, they analysed the inflammatory breast cancer cells using a kinase antibody assay and revealed that only co-culture with M2 macrophages can these tumour cells elevate kinase C zeta (PRKCZ) level, leading to radiation resistance in radiotherapy (Ref. Reference Rahal145). Another study also demonstrated that SEPT9 which mediated M2 macrophages polarisation increases irradiation resistance by interacting with the HMGB1-RB axis (Ref. Reference Jiao146) (Fig. 2 green box). However, the irradiation dose can influence the infiltration subpopulation of TAMs. Low doses (below 1 Gy) and high dose (>10 Gy) evoke low toxicity and favour the M2 phenotype of TAMs whereas intermediate dose (1–10 Gy) induces a shift of unpolarised macrophages toward the M1 phenotype (Ref. Reference Malfitano147). Therefore, it is important to choose an appropriate dose to minimise the resistance to radiotherapy while eliminating tumours.
When it comes to origins, Akkari et al. demonstrated that the ratio of TRMs and tumour-infiltrating BMDMs was altered during radiotherapy response and recurrence. In radiotherapy-recurrence gliomas, BMDMs were increased compared to untreated controls, constituting ⩾50% of total TAMs, and inhibition of BMDMs infiltration delays glioma recurrence after radiotherapy. When blocking both TRMs and BMDMs using BLZ945, a blood-brain barrier–permeable CSF-1R tyrosine kinase inhibitor to specifically target these TAM populations together, tumour regression was associated with increased apoptosis and decreased glioma cell proliferation in the combination treatment group compared to each monotherapy (Ref. Reference Akkari83). This study reveals that both TRMs and BMDMs limit the efficacy of radiotherapy and support glioma recurrence after radiotherapy. However, other studies hold the view that TRMs display radiation resistance whereas BMDMs did not resist irradiation. Although both subsets upregulate the Cdkn1a gene, only TRMs exhibit a greater increase after irradiation. And the radio-resistance is compromised in the absence of the Cdkn1a gene and its protein product, p21cip1/WAF1, further confirming the important function of the Cdkn1a gene and p21cip1/WAF1 protein in radio-resistance (Ref. Reference Soysa148).
Immunotherapy resistance
According to previous studies, we classify this immunotherapy associated with macrophages into five main groups: (1) inhibiting recruitment of monocytes, such as CSF1/CSF1R inhibitor and CCL2/CCR2 inhibitor; (2) blocking pro-tumour functions, such as VEGFR inhibitor; (3) promoting phagocytosis of macrophages, such as CD47/SIRPα inhibitor; (4) reprogramming TAMs into an immunostimulatory phenotype, such as CD40 agonists; (5) inhibiting immune checkpoint proteins, such as PD-L1 inhibitor and CTLA4 inhibitor. To date, numerous therapeutic strategies specifically modulating TAM functions have shown promising results in preclinical studies and clinical trials (Table 1). However, the tumour recurrence rate of various tumours is still high, bringing a huge problem, immunotherapy resistance, to clinical therapy.
Table 1. Immunotherapy associated with macrophages
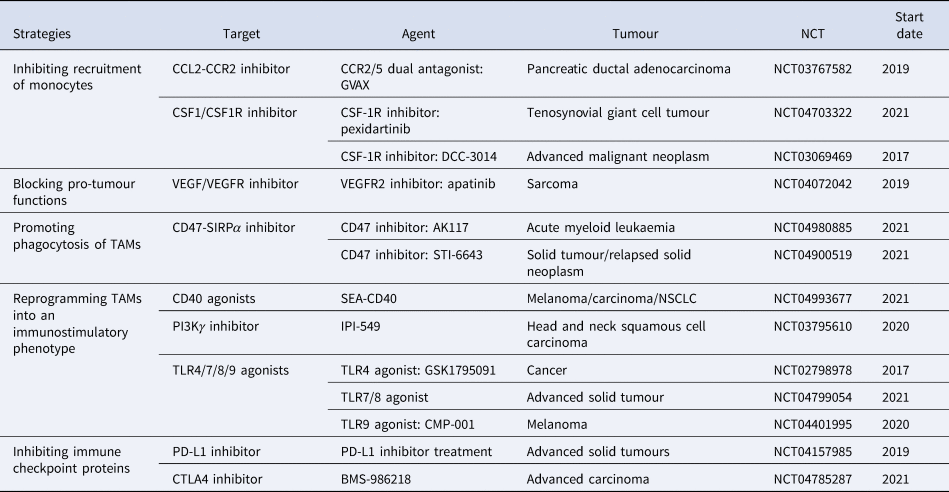
Data were obtained from http://clinicaltrials.gov.
Although CSR-1R inhibitors prolonged the OS of glioblastoma multiforme in mouse models, more than 50% of tumours eventually recurred accompanied with elevated phosphatidylinositol 3-kinase (PI3K) pathway activity (Ref. Reference Quail149). In recurrence tumours, the IGF-1 which is upregulated in TAMs through the IL4-NFAT pathway is secreted into the extracellular environment. Then, IGF-1R and PI3K signalling pathways are activated in glioma cells, supporting tumour growth and malignancy.
In the immunotherapy resistance murine cancer models to anti-VEGF antibody, they found upregulated alternative angiogenic pathways in VEGFR-1 and VEGFR-3 depletion macrophages, facilitating escape from anti-VEGF therapy (Ref. Reference Dalton150). Therefore, finding out alternative angiogenic pathways and targeting them is essential for immunotherapy resistance. When employing late-stage RIP1-Tag2 pancreatic neuroendocrine tumours (PNETs) which develop resistance to VEGFR2 blockade, Rigamonti et al. found upregulated ANG2 and TIE2 accompanied with enhanced infiltration of TIE2+ macrophages. Further study found that dual ANG2/VEGFR2 blockade inhibited revascularisation and progression in most of the PNETs (Ref. Reference Rigamonti151).
Another immunotherapy, anti-CD40 treatment, upregulated the expression of PD-L1 on tumour-infiltrating monocytes and macrophages, indicating immunotherapy resistance. To reverse this condition, both anti-CD40 antibody and PD-L1 blocking antibody were applied in murine tumour cell lines. This double-antibody therapy resulted in complete tumour rejection in >50% of the mice, whereas anti-CD40 treatment alone only led to <5% tumour rejection in mice (Ref. Reference Zippelius152). Until now, the expression of PD-1/PD-L1 within the TME is considered as the main mechanism escaping immune surveillance and enhancing resistance, uncovering an idea that the blockade of the PD-1/PD-L1 axis is an available therapeutic strategy against resistance (Refs Reference Peng153, Reference Gong154) (Fig. 2 pink box). Interestingly, a study pointed out that, except membrane-localised PD-L1, a portion of PD-L1 is also located in the nucleus and cytoskeleton in human and mouse cancer cell lines (Ref. Reference Gao155). Moreover, blocking its nuclear translocation through acetylation enhanced the anti-tumour response to PD-1 blockade, indicating that the location of PD-L1 is of great importance in resistance (Ref. Reference Fang156). Thus, we propose a question: whether the PD-L1 location in macrophages also changed during immunotherapy resistance? (Fig. 3).
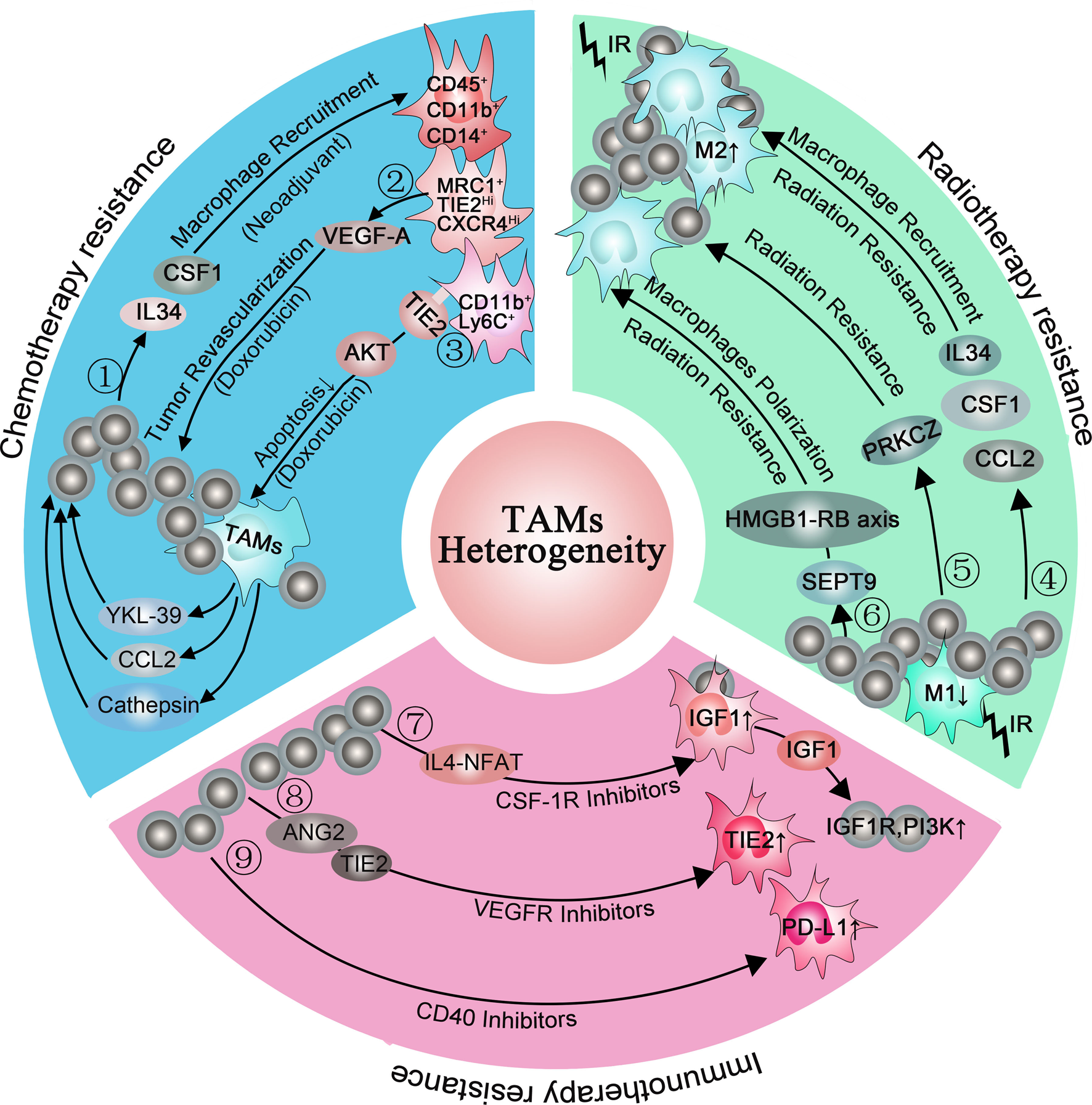
Fig. 3. Macrophages are involved in chemotherapy resistance (blue box), radiotherapy resistance (green box) and immunotherapy resistance (pink box). ①: Chemotherapy (neoadjuvant) induced mammary epithelial cells to produce macrophage recruitment factors, including colony-stimulating factor 1 (CSF1) and interleukin-34, and a higher percentage of CD45+CD11b+CD14+ macrophages infiltrated in breast cancer biopsy samples. ②: (MRC1+ TIE2Hi CXCR4Hi) macrophages promoted tumour revascularisation and relapse after chemotherapy (Doxorubicin) via VEGF-A release. ③: CD11b+ Ly6C+ macrophages abundantly expressed TIE2 and prevented macrophages from apoptosis via the AKT-dependent signalling pathway. ④: Radiotherapy increased the expression of macrophage recruitment factors, including colony-stimulating factor 1 (CSF1), CCL2 and interleukin-34, promoting M2 macrophages polarisation. ⑤: M2 macrophages elevated the level of PRKCZ in the tumour and leading to radiation resistance in the end. ⑥: SEPT9 increases irradiation resistance by interacting with the HMGB1-RB axis. ⑦: CSF-1R inhibitors elevated the IGF1 protein level in macrophages via the IL4-NFAT pathway, then IGF-1 secreted into the extracellular environment and resulted in activation of IGF-1R and PI3K signalling in tumour cells. ⑧: VEGFR inhibitors elevated TIE2 protein level in macrophages via ANG2, upregulating angiogenic pathways. ⑨: CD40 inhibitors increased PD-L1 expression in macrophages.
Conclusion and prospect
TAMs are highly plastic cells infiltrated in tumours. These macrophages are related to tumour death, tumour initiation, migration and invasion. TAMs heterogeneity increases resistance in cancer therapy, emphasizing the importance of defining the subpopulation of TAMs in the TME. Macrophages are divided into TRMs and BMDMs according to their sources, divided into M1 and M2 macrophages according to different surface molecules, and subdivided into different subpopulations according to their function. Additionally, more classification methods of macrophages are being discovered recently. However, before the relationship between macrophages and tumours can be fully understood, many questions remain to be answered: (1) Is there any preference for polarisation pathways in different tumours? (2)which classifications cover all types of macrophages? (3) which subpopulation can provide the most suitable target for clinical therapy? (4) why can macrophages differentiate into different subsets? (5) what are the consequence of increased macrophages heterogeneity?
Macrophage heterogeneity is related to tumour heterogeneity, stimulating factor heterogeneity and TME heterogeneity. Among them, tumour heterogeneity is mainly manifested as (1) the functions of macrophages from different sources are different; (2) the localisation of macrophages in the same tumour is different; (3) there are differences in macrophages in different types of tumours. The heterogeneity of stimulating factors is mainly manifested by the difference in stimulation pathways. M1 macrophage polarisation is mainly involved in the LPS/IFN-γ polarisation pathway, while M2 macrophage polarisation is involved in the IL4/13/10 polarisation pathway. In addition, non-coding RNA also participates in macrophage polarisation.
Because of the different sensitivity of macrophages to therapy, the number of macrophages that are resistant to chemotherapy, radiotherapy and immunotherapy eventually increases, which in turn causes tumour recurrence. Therefore, studying the subpopulations that increased in therapy resistance and targeting them will guide clinical therapy in the future.
This review provides diverse introductions to TAM heterogeneity. From a clinical point of view, we provide the possibility to precisely target different subpopulations of TAMs.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81971341; 82101704) and China Postdoctoral Science Foundation (No. 2020M682674; 2021T140144).
Conflict of interest
None.





 Open access
Open access