


INTRODUCTION
Giardia lamblia (syn. G. duodenalis or G. intestinalis) is a human and animal intestinal protozoan parasite and is a frequent cause of diarrhoea. Repeated or chronic infection is common in areas of poverty and overcrowding [Reference Gilman1]. In affluent countries, rates are lower, but when hygiene breaks down (e.g. in children's nurseries, or in association with travel abroad, camping and recreational water contact, or as a result of contaminated drinking water systems) rates can be similar to those seen in developing countries [Reference Pickering2–Reference Gray and Rouse4].
Molecular techniques developed in the 1980s and 1990s have revealed a rich diversity of genetic subtypes within the species G. lamblia [Reference Ey5–Reference Monis7]. As currently understood, there are two main genogroups or genetic assemblages of G. lamblia which infect humans – Assemblage A and Assemblage B [Reference Caccio and Ryan8]. These genogroups are also found in animals, and have been divided into subassemblages: AI, AII, BIII and BIV. A further subassemblage (AIII) has recently been described, but only in animals [Reference Caccio9]. Other genogroups also affect animals, but are rarely or never found in humans: genogroups C and D in dogs [Reference Hopkins6], genogroup E in ruminants [Reference Ey5], and genogroups F and G in cats and rats, respectively [Reference Monis7].
Assemblages A and B express different surface proteins [Reference Ey and Darby10], and differ in their ability to be cultivated in vitro [Reference Karanis and Ey11]; recent genomic studies suggest that the genetic differences are so great that they should be regarded as different species [Reference Franzen12]. These differences could lead to differences in the organisms' ability to spread and cause disease, although the degree to which this occurs is uncertain. The purpose of this study was to examine cases of giardiasis in an urban cosmopolitan setting in the UK, to determine the prevalence of different genetic subtypes and whether these subtypes differed in terms of clinical or epidemiological features.
MATERIALS AND METHODS
Clinical samples and surveillance data
Cases of microscopically confirmed giardiasis occurring in south-west London between 1999 and 2005 were studied. Most cases were symptomatic; a small proportion (<2%) were detected when asymptomatic contacts of symptomatic cases were screened.
The three hospital diagnostic microbiology laboratories in the area (St George's, St Helier's, Kingston hospitals) were asked to provide faecal samples that had been found to contain Giardia cysts. Two laboratories used direct microscopy, and one laboratory used a commercial concentration procedure (Parasep™, DiaSys, UK) before microscopy. Samples were stored (not formalinized) at 4°C until analysis.
The five environmental health departments in the area (Kingston-upon-Thames, Richmond-upon-Thames, Merton, Sutton, Wandsworth) were asked to provide surveillance data on cases of giardiasis that had been notified to them. These departments investigate giardiasis using a standard postal questionnaire. These surveillance data were combined with demographic information included on the laboratory request form.
Descriptive statistical methods were employed, using an electronic spreadsheet program (Microsoft Excel version 9.0). Tests of significance included Fisher's exact test, the Mann–Whitney U test, and where appropriate, 95% confidence intervals.
DNA extraction and purification
Whole faecal DNA extraction and purification was performed using the Boom method [Reference Boom13] as modified by McLauchlin et al. [Reference McLauchlin14]. In brief, 200 mg faecal samples were homogenized with 0·5 mm zirconia/silica beads, to disrupt cysts, with isoamyl alcohol to reduce foaming. DNA was extracted with guanidine isothiocyanate (GuSCN), with binding to silica beads. Following removal of denatured proteins, fats and other faecal material by repeated washing with GuSCN buffer, alcohol and acetone, the DNA was eluted with 150 μl water. Samples which were negative by initial PCR were further purified with polyvinyl pyrrolidone (PVP) to remove residual PCR inhibitors [Reference Lawson15]. Control DNA was from two axenic strains of G. lamblia: PO-1 [Reference Meyer16] and NESS, a strain isolated from a human case in Scotland (H. V. Smith, Scottish Parasite Diagnostic Laboratory, personal communication).
Sequenced-based typing
Sequence-based typing at the ribosomal RNA gene (rRNA) and triose phosphate isomerase gene (tpi) loci was performed by the following method. Candidate gene sequences were retrieved from the Genbank sequence database [17]. Multiple sequences for the same genes were aligned using the ClustalW program, available from the website of the European Bioinformatics Institute (EBI) [18]. Where possible, gene sequences representing divergent species of Giardia were included, including G. lamblia, G. ardeae, G. muris and G. microti, in order that genus-specific primers could be selected. Candidate primers were selected from conserved areas, and checked for compatibility and stability using the Netprimer program [19].
Primer oligonucleotides (Table 1) were synthesized by Invitrogen Corp. (USA). These were suspended in distilled water to a concentration of 50 pmol/μl. PCR reaction mixtures were prepared with 10 μl faecal DNA extract and 40 μl mastermix, with a final concentration of 1×PCR buffer (Invitrogen), 0·2 mm dGTP, 0·2 mm dCTP, 0·2 mm dATP, 0·2 mm dTTP, 1·5 mm MgCl2, 1 μm of each primer, 2·5 U Taq polymerase (Platinum™, Invitrogen Corp.) per reaction, 0·2× Q-solution (Qiagen GmbH, Germany). The mastermix was prepared on ice, and nested or hemi-nested PCRs were performed with a hot-start protocol: 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 55°C for 2 min and 72°C for 3 min, finishing with 7 min at 72°C. For the second-round reaction, 10 μl of a 1/20 dilution of the first-round product was processed using the same conditions. PCR products were detected by gel electrophoresis.
Table 1. Primers used in the rRNA and tpi PCRs

T m, Melting temperature; GC, guanine-cytosine content.
* Key to degenerate nucleotides in primer sequences: m=a/c; r=a/g; s=g/c; y=c/t.
† Primer tpi1f was used as the forward primer in both rounds of a hemi-nested reaction.
Predicted rRNA gene PCR product sizes, in base pairs: 847 (outer), 506 (inner).
Predicted tpi gene PCR product sizes: 518 (outer), 339 (inner).
Sequencing was performed by Qiagen GmbH using the Sanger chain termination method [Reference Sanger, Nicklen and Coulson20]. Sequencing was done in both the forward and reverse directions. ClustalW was used to align sequences with comparator Giardia sequences of known assemblage. Phylogenetic trees of these alignments were generated using two methods: the neighbour-joining method [Reference Saitou and Nei21], using the ClustalX program, available from the Clustal website [22] and a maximum parsimony algorithm within the PHYLIP set of programs [Reference Felsenstein23]. The trees were visualized using NJPlot, a tree-drawing program available from Pôle Bio-Informatique Lyonnais [Reference Guoy24], and bootstrap analysis was performed using PHYLIP. Sequences were assigned to Assemblages A or B according to which Genbank sequence they most closely aligned with.
Duplex PCR-based typing
This was a hemi-nested PCR for tpi, as described previously [Reference Amar25]: the first round was duplex, with separate primers for Assemblages A and B; the second round consisted of separate PCR reactions for each assemblage. As each reaction yielded different sized fragments (Assemblage A product 452 bp, Assemblage B product 141 bp), gel visualization of the PCR product could distinguish the two assemblages. This method also enabled mixed infections of Assemblages A and B to be detected.
Sequence data accession numbers
DNA sequences from this project are available in Genbank [17]: tpi accession numbers FM242108–FM242136; rRNA gene accession numbers FN252252–FN252290.
RESULTS
Demographics of patients
Between 1999 and 2005, 819 cases of giardiasis were diagnosed in the three laboratories in south-west London. Environmental health questionnaires were returned by 389/819 cases.
The age distribution was bimodal, with peaks in infancy and in adults in their 30s (Fig. 1). The sex distribution was 47·7% male, 52·3% female. Outbreaks (as defined by two or more linked cases) were common, with 101 separate outbreaks reported involving 170 (21%) of the cases; but 95 of these outbreaks were small, single household clusters: the mean number of study cases in each outbreak was 1·6.
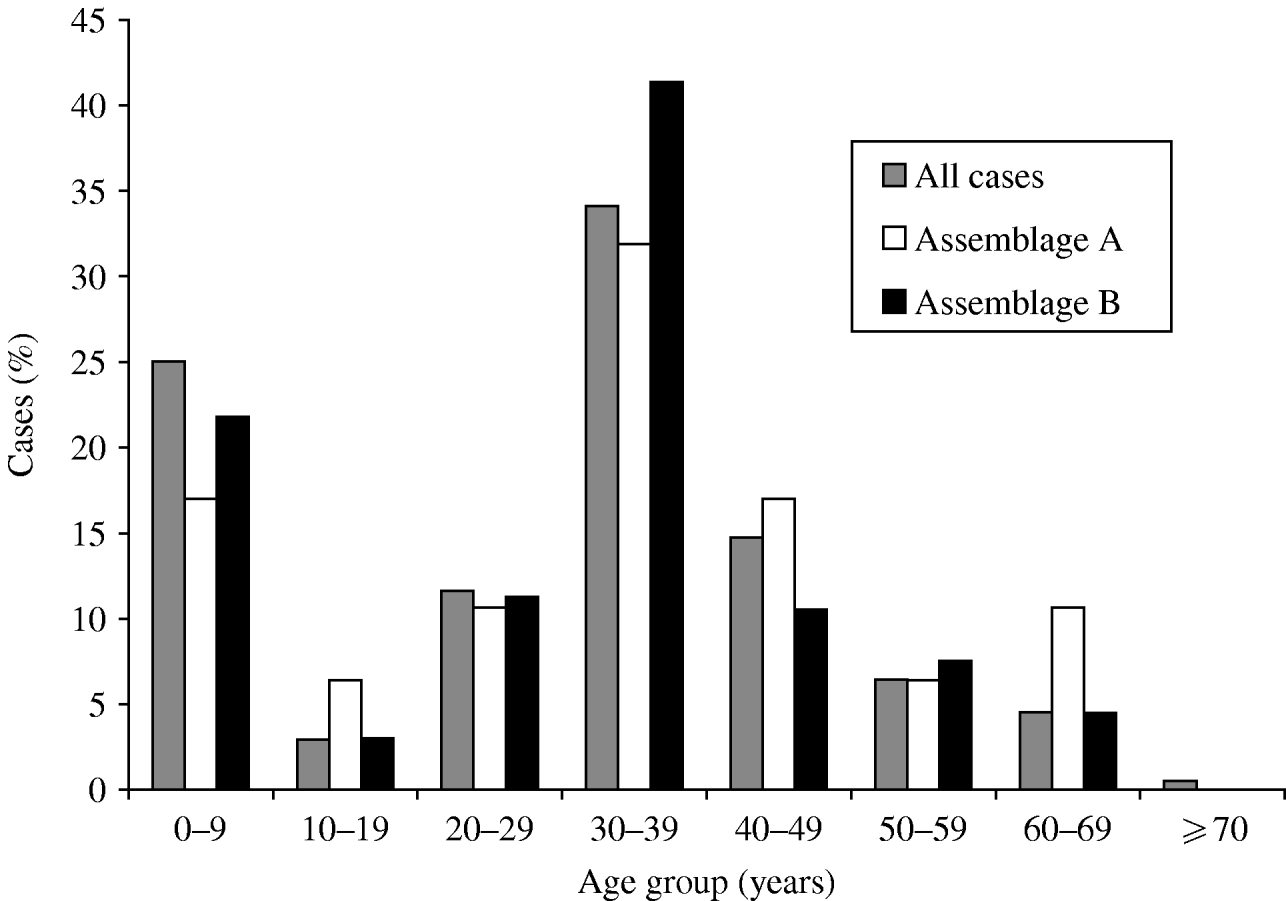
Fig. 1. Age distribution of cases of giardiasis, for all cases where age was recorded (n=747), and Assemblage A (n=47) and Assemblage B (n=133). Mixed infections excluded.
Recent travel was recorded in 208/389 (53%) questionnaire responses; 104 (27%) patients indicated they had not travelled and the remainder left the question unanswered.
Stool sample selection for molecular analysis
Of the 819 identified cases, 574 had samples submitted for this study. Only 144 cases had both samples submitted and questionnaires returned. Samples that had clinical and/or epidemiological information available were favoured for molecular analysis. On this basis, 267/574 available stool samples were selected for DNA extraction and PCR analysis (Fig. 2).
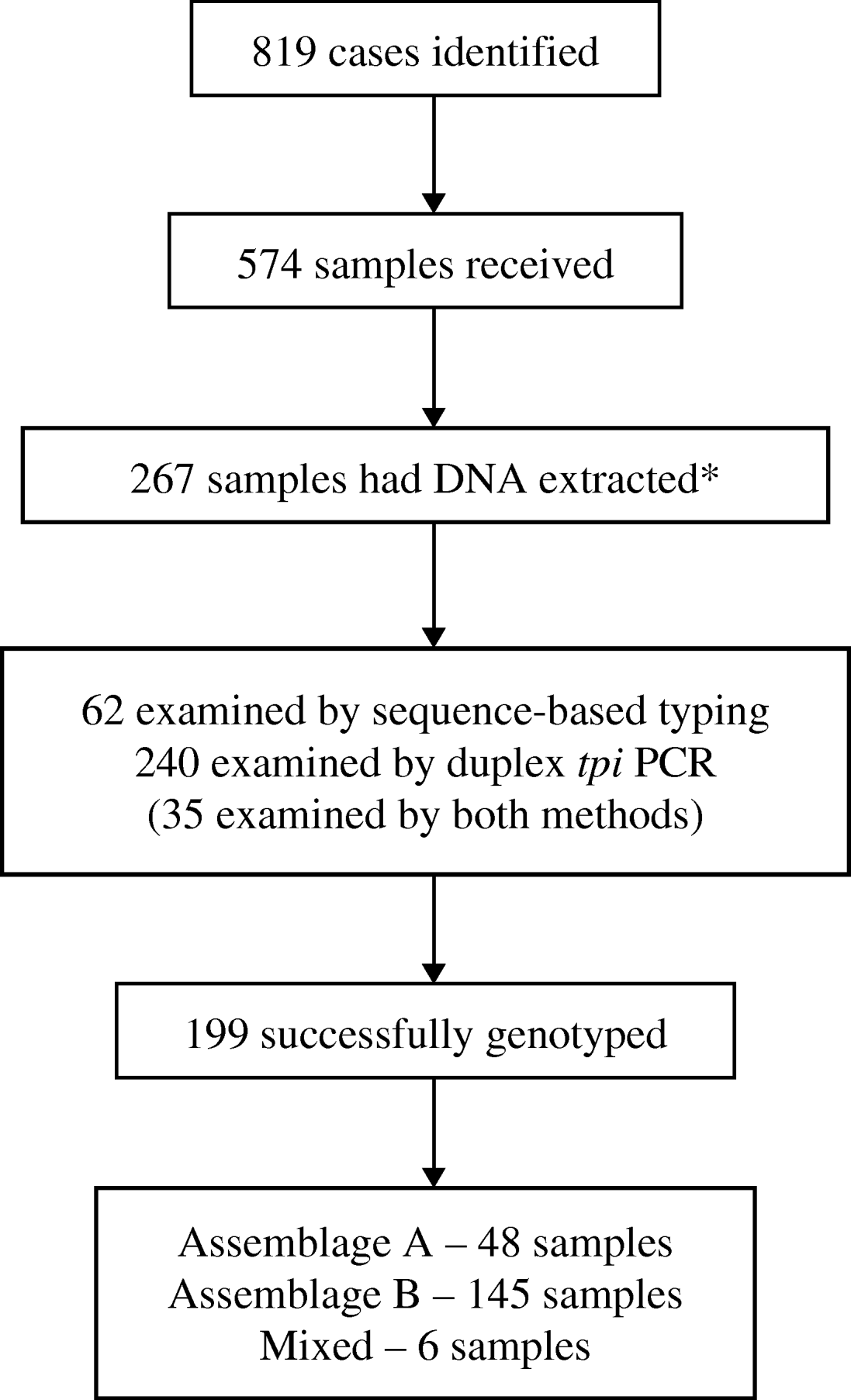
Fig. 2. Flow chart showing numbers of cases of giardiasis identified and samples received, and numbers subsequently analysed and successfully genotyped. (* Samples with epidemiological or clinical information were preferred for molecular analysis.)
Sequence-based typing
PCR for the rRNA and tpi genes was performed on 62 DNA extracts. Products of the predicted size were obtained from 41/62 rRNA and 48/62 tpi PCR reactions. From these PCR products 39 rRNA gene sequences and 28 tpi sequences were obtained. A positive sequence for one or both genes was obtained from 45 samples.
Phylogenetic trees generated from the sequence data showed that all clinical samples clustered with either Assemblage A or Assemblage B; there were no intermediate clusters (Fig. 3). Different tree-generating algorithms, as well as bootstrapping, were used in order to assess the reliability of any inferences being made. Clustering was concordant between the two genes for all samples, and the trees generated by both neighbour-joining and maximum parsimony methods were similar. It was possible to assign 14 (31%) isolates to Assemblage A, and 31 (69%) to Assemblage B, by one or both sequencing reactions.
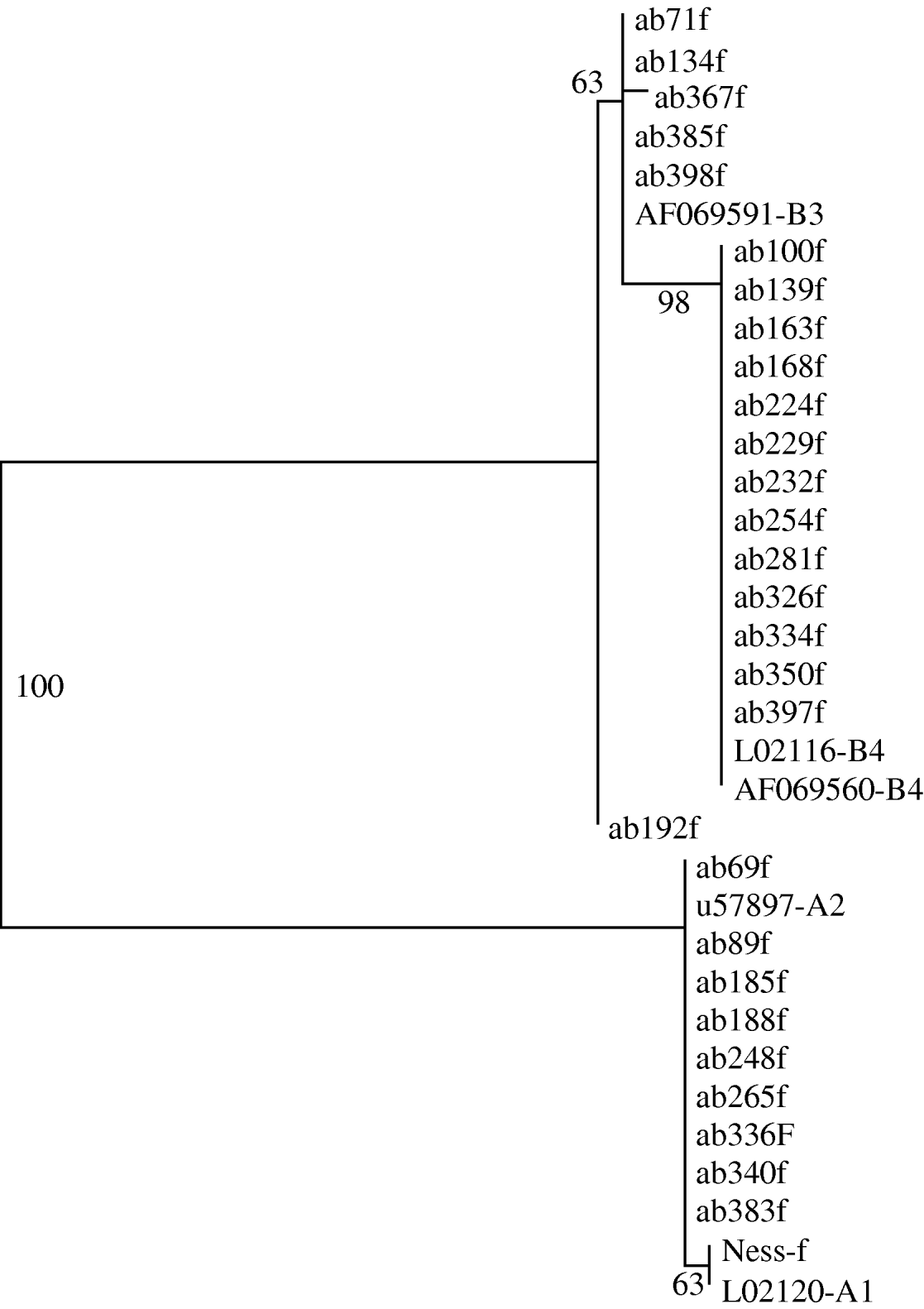
Fig. 3. Phylogram derived from forward tpi sequences, using a maximum parsimony algorithm. Genbank reference sequences of known assemblages are included. Bootstrap values are shown, from 100 iterations. Ab192f differed from Assemblage B3 by just 1 base pair.
The rRNA gene sequences did not reveal any subassemblages, but the tpi sequences had subgroups which aligned with known Assemblage BIII and BIV sequences (Fig. 3). The tpi sequences of all Assemblage A clinical samples clustered with Assemblage AII – none aligned with AI. The tpi sequences showed most variation between the two assemblages, at 57/246 bp aligned (23%), compared to only four differences out of 431 aligned base pairs for the rRNA gene sequences (0·9%).
Duplex PCR-based typing
Duplex PCR-based typing was performed on 240 DNA extracts. Of these, 35 had already been typed by sequencing, to ensure concordance between the two methods. Molecular typing was successful for 164 (70%) samples. Assemblage A accounted for 36 (22%) samples and Assemblage B for 125 (75%); six (4%) samples were mixed.
Combined typing results
In total, 267 samples were examined by at least one of the PCR methods, including 35 that were tested by both methods. Genetic typing was successful by at least one method for 199 (75%) samples. Assemblage A accounted for 48 (24%) samples; Assemblage B for 145 (73%), and six (3%) were mixed. Where samples were successfully typed by both methods, the results were concordant in all cases.
Clinical and epidemiological correlations with Assemblages A and B
Assemblages A and B had similar patient demographics. Both assemblages had a bimodal age distribution. Males accounted for 44% of Assemblage A cases, and 49% of Assemblage B cases (P=0·6159, Fisher's exact test). Both assemblages had similar proportions of cases associated with household outbreaks: 6/49 (18%) Assemblage A cases, and 33/148 (22%) Assemblage B cases (P=0·358). In nine outbreaks (all family clusters), two or more samples were successfully genotyped: in seven of these, the assemblages were concordant, but in two outbreaks Assemblages A and B were detected.
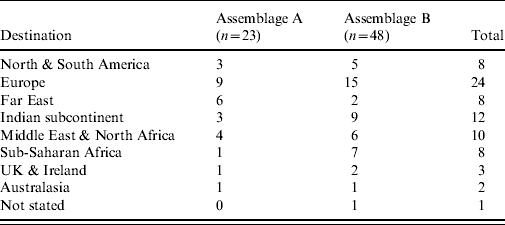
Travel histories were available for 98 of the cases where molecular typing was successfully performed. The proportions of Assemblage A in recent travellers (n=73) and non-travellers (n=25) were similar (32% and 33%, respectively; P=0·56). The travel-associated cases had visited a wide number of destinations (Table 2); the numbers for each destination were too low for meaningful statistical analysis.
Table 2. Destinations of travel-associated giardiasis cases by assemblage (n=71). Two infections with mixed Assemblages A and B are excluded (note that some cases had visited more than one destination)
Both assemblages peaked in incidence at the end of the year (October–December) (Fig. 4). The duration of illness before diagnosis was known for 91 cases, and ranged widely for both assemblages, from 4 or 5 to 180 days. Assemblage A had a shorter mean duration of illness (25·8 days, compared to 26·6 days for Assemblage B), and a greater proportion of Assemblage B cases (44%, compared to 21% of Assemblage A) had a prolonged illness (⩾20 days). However, the differences between the two assemblages did not reach statistical significance (P=0·17, Mann–Whitney U test).
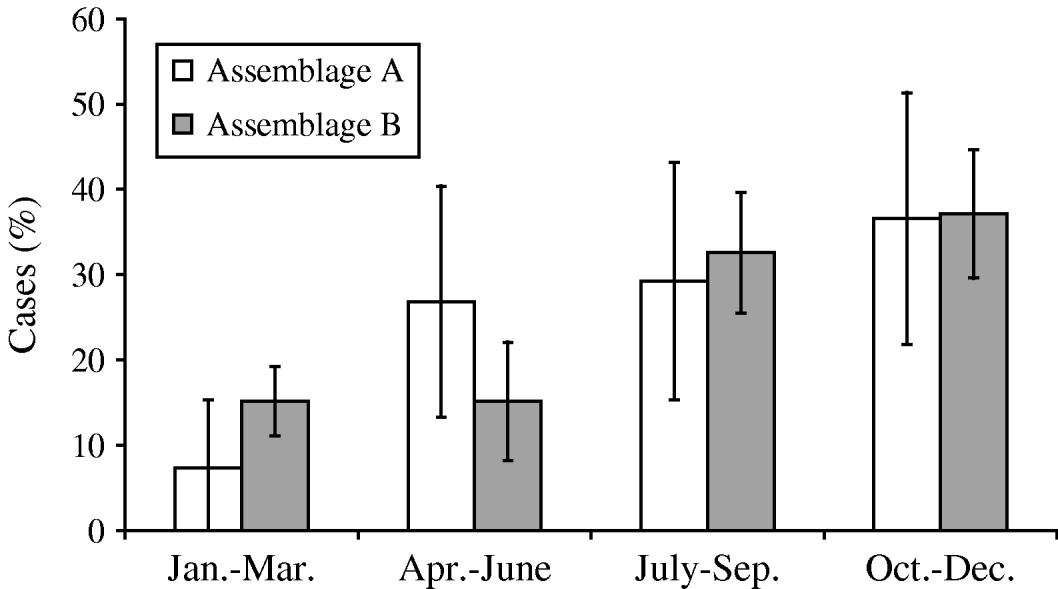
Fig. 4. Seasonal distribution of Assemblages A and B cases.
Fifty-nine of the patients whose isolates had been successfully genotyped provided details of symptoms (Table 3). Fever was significantly more common in Assemblage A than Assemblage B [odds ratio (OR) 5·091, 95% confidence interval (CI) 1·39–18·81; P=0·018 by Fisher's exact test]. For all other symptoms, there were no significant differences.
Table 3. Symptoms in cases of Assemblages A and B
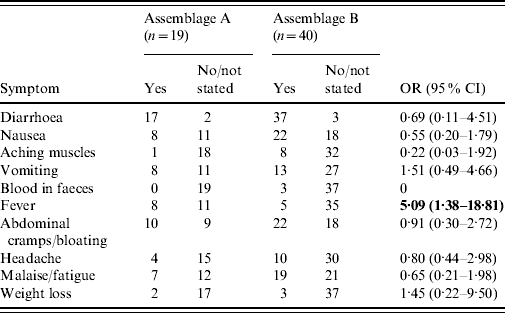
OR, Odds ratio; CI, confidence interval.
Bold values indicate statistically significant values.
Patients with Assemblage A organisms reported more symptoms (mean 2·88) than those with Assemblage B (mean 2·29), although this is not statistically significant (P=0·1738, Mann–Whitney U test). Of note, three patients with no symptoms, who were picked up on screening of contacts of cases, were all infected with Assemblage B.
DISCUSSION
This study shows the proportions of G. lamblia Assemblages A and B in a cosmopolitan urban UK population. Both assemblages have similar epidemiological and clinical features, but there are differences between assemblages in terms of presence of fever.
Methodological approach
PCR applied directly to clinical samples was used. Two housekeeping genes were chosen for PCR analysis, in order to enhance robustness of typing. A quarter of samples failed to amplify by either PCR assay, despite PVP purification. Probable explanations are persisting PCR inhibitors in the samples, and/or low numbers of cysts (cyst numbers were not quantified before PCR). The tpi gene PCR products did not yield a sequence in 20/48 cases, compared to only 2/41 rRNA gene PCR products which failed to sequence. The reason for this difference is uncertain. The tpi PCR reactions were noted to have a higher proportion of non-specific bands, and the gene is common to most faecal bacteria, so it is possible that some of the positive PCR results included products of non-specific amplification, which could have interfered with the sequencing process.
Sequence-based typing for the tpi gene allowed differentiation of strains within the two major assemblages, into the four recognized subassemblages already described (AI, AII, BIII, BIV – although A1 was absent from the clinical isolates). Sequencing may miss mixed infections with different assemblages in a single host (subsequently shown to be <5% by duplex PCR). Sequencing may also miss heterozygosity between the nuclei in a binucleate organism. The Assemblage A genome sequencing project has estimated heterozygosity in the genome of the strain studied (the WB clone C6) to be <0·01% [Reference Morrison26], but the Assemblage B genome has an estimated heterozygosity of 0·53%, which could explain some single-nucleotide differences between strains within Assemblage B [Reference Franzen12].
The duplex PCR-based typing method was adopted when the initial sequence results showed the limited genetic diversity within Assemblages A and B, and the absence of infections due to other genogroups. This method had the advantage of relative ease and faster throughput, and also avoided the loss of data that occurred with the extra sequencing step.
Preponderance of Assemblage B
The typing results show that all Giardia infections in this population are due to G. lamblia Assemblages A and B. This confirms the results of a number of studies performed elsewhere [Reference Caccio and Ryan8, Reference Karanis and Ey11, Reference Lalle27].
The preponderance of Assemblage B strains in this London population (73%) is similar to the results of a smaller British study which showed that 21 (64%) of 33 Giardia isolates were Assemblage B, with 9% mixed Assemblages A and B [Reference Amar25]. The worldwide distribution is less consistent, with a reported proportion of Assemblage A ranging from 0% in a small number of Indian isolates [Reference Sulaiman28] to 100% in studies from Central America and Portugal [Reference Eligio-Garcia, Cortes-Campos and Jimenez-Cardoso29, Reference Sousa30]. However, molecular studies of Giardia may not be directly comparable: some studies are limited to particular outbreaks [Reference Robertson31], whereas other studies use diagnostic samples from symptomatic patients [Reference Sousa30], case-control studies [Reference Haque32], and surveillance studies of particular cohorts of patients. These studies also use many different primers and molecular-typing methods, and it is possible that different methodologies may favour one assemblage over another [Reference Lu33].
Although outbreaks were common in our study, these were mostly small family clusters of cases; there were no large outbreaks which could potentially have skewed the overall results. Outbreak- and non-outbreak-associated cases had similar proportions of each assemblage. Of note, 2/9 outbreaks had mixed assemblages detected, which serves as a reminder that, while some outbreaks may represent cross-infection from one linked case to another, others may represent shared exposure to unhygienic conditions, where mixed assemblages may be more likely.
Differences between Assemblages A and B
Given the genetic and phenotypic differences between assemblages, it might be expected that there would be correlating clinical/epidemiological differences. In fact, in most of the parameters examined Assemblages A and B caused broadly similar illness, although the number of patients with both genotype and symptom data was low, and only one statistically significant difference was noted: fever was significantly more common in Assemblage A. Other differences which did not reach statistical significance included a trend towards a higher number of symptoms in Assemblage A, and a longer illness or asymptomatic infection in Assemblage B. Several other studies which examined clinical differences between assemblages support the hypothesis that Assemblage B is associated with persistence or asymptomatic carriage [Reference Haque32, Reference Aydin34–Reference Sahagun37]. Such a tendency may explain discrepancies between the prevalence of Assemblages A and B in returned travellers, compared to local genetic typing studies carried out in the destination of travel; for example, our study found 3/4 (unlinked) cases in travellers returned from Portugal to be due to Assemblage B, whereas a study carried out in Portugal found all of 25 symptomatic cases to be due to Assemblage A [Reference Sousa30].
However, other work has found greater proportions of asymptomatic infection in Assemblage A [Reference Gelanew38]. A small Dutch study (n=18) looked only at symptomatic cases in some detail [Reference Homan and Mank39]: they found that the nine Assemblage B cases all had active diarrhoea at the time of diagnosis, whereas the nine Assemblage A cases all had intermittent and short-lived diarrhoea. Contrary to the present study, they also found that Assemblage B cases were more likely to present with fever, and had more severe illness than Assemblage A cases. On the basis of their findings, they suggested that Assemblage A infection is more likely to lead to asymptomatic carriage – an hypothesis not supported by the present study or other studies [Reference Haque32, Reference Aydin34–Reference Sahagun37].
This study confirms the predominance of Assemblage B in Giardia cases in an urban British setting, in both travel-associated and non-travel-associated cases. Both assemblages had similar clinical and epidemiological features, although Assemblage A infections more commonly presented with fever, and had non-significant trends towards a greater number of reported symptoms and a shorter duration of illness. Further work is necessary to definitively characterize clinical differences between the assemblages.
ACKNOWLEDGEMENTS
This work was supported by a grant from the Trustees of St George's Hospital. We thank the following: environmental health officers in Merton, Sutton, Wandsworth, Kingston-upon-Thames and Richmond-upon-Thames, for their assistance in allowing access to routine surveillance data of cases. Laboratory staff in St George's, Kingston and St Helier's hospitals, for providing clinical faecal samples. Dr C. F. Amar (Centre for Infections, Health Protection Agency, London), for her generous advice and guidance in helping us perform duplex PCR genotyping of isolates.
DECLARATION OF INTEREST
None.







