


Fe deficiency is the most common nutrient deficiency worldwide and is particularly prevalent in children, women of child bearing age and pregnant women. Even in the UK, it is estimated that 23 % of women aged 19–64 years and nearly half of teenage girls do not reach their lower reference nutrient intake for Fe( Reference Miller, Spiro and Stanner 1 ). During development, the fetus relies entirely on maternal supply for Fe, Zn and Cu, which are critical for development and health. Crosstalks between these three trace elements have been established decades ago( Reference Collins, Prohaska and Knutson 2 – Reference Whittaker 4 ), but the effect of Fe deficiency on the metabolism of Cu and Zn is still not fully understood, particularly during pregnancy. The ability to carry out work establishing these links is, of course, limited, but micronutrient metabolism is well-conserved in mammals, and the rat has been shown to be a very good model for pregnancy and nutrition in humans( Reference Gambling, Kennedy and McArdle 3 ).
Generally, Fe deficiency in mammals results in increased Cu levels in the blood, gut and liver( Reference Owen 5 – Reference Sourkes, Lloyd and Birnbaum 7 ). Circulating ceruloplasmin (CP), produced by the liver, and its gut homologue hephaestin are Cu-dependent ferroxidases and appear as an evident link between Fe and Cu metabolism. Accordingly, Fe deficiency in rats induces an increase in serum Cu associated with an increase in CP levels( Reference Owen 5 ). Interestingly, Fe-deficient rats have higher liver Cu levels that correlate with CP expression and its ferroxidase activity, while hepatic CP mRNA remains unchanged, suggesting that Cu loading is associated with an increased metallation of the protein( Reference Ranganathan, Lu and Jiang 8 ). Fe deficiency also increases the expression of the Menkes Cu ATPase (ATP7a) in the duodenum( Reference Ravia, Stephen and Ghishan 6 , Reference Collins, Franck and Kowdley 9 ), suggesting an increased export of Cu from the enterocyte to the circulation, while Wilson Cu ATPase (ATP7b) expression in the liver is unaltered( Reference Ravia, Stephen and Ghishan 6 ). In addition, Cu and Fe may compete for divalent metal transporter 1 (DMT1) for their uptake in the gut. Although Cu influx through DMT1 in the enterocyte is minor compared with Cu transporter 1 (CTR1) in replete conditions, it is of particular importance during Fe deficiency due to its up-regulation and the lack of Fe in the lumen, which favours Cu uptake( Reference Jiang, Garrick and Garrick 10 ).
The effect of Fe deficiency on Zn status is believed to be less important than on Cu status. Fe deficiency appears to reduce Zn requirements in rats, increasing Zn levels in several organs such as liver, spleen, brain or kidney( Reference Shukla, Agarwal and Shukla 11 , Reference Shukla, Agarwal and Shukla 12 ) while decreasing apparent Zn absorption( Reference Kaganda, Matsuo and Suzuki 13 ) and plasma Zn( Reference Yokoi, Kimura and Itokawa 14 ). Noteworthy, marginal Fe deficiency (9 and 13 mg Fe/kg diet) did not affect Zn status in rat, while it was sufficient to induce Cu accumulation in the liver( Reference Stangl and Kirchgessner 15 ). Because Fe and Zn have similar mechanisms of absorption and transport in the gut, they were originally thought to compete with each other, notably through DMT1. Accordingly, Fe treatment reduces the transport of Zn in Caco-2 cells although to a lesser extent than other divalent cations( Reference Tallkvist, Bowlus and Lonnerdal 16 ). However, DMT1’s affinity for Zn is much lower than that for Fe, and its contribution to Zn transport and uptake is probably minimal( Reference Illing, Shawki and Cunningham 17 ).
Two families of Zn transporters (ZnT) have been identified, ZRT/IRT-like protein (ZIP) and ZnT, generally involved in influx and efflux of the cells, respectively. ZIP stands for Zn-regulated transporter, Fe-regulated transporter-like protein, and is composed of 14 members in mammals. They are characterised by their ability to transport Zn, but several of them have also been reported to mediate the uptake of Fe as well as Mn( Reference Guerinot 18 ). In rats, Fe overload increases the expression of several ZIP in the liver, namely, ZIP5 (up), ZIP6, ZIP7 and ZIP10 (down), while ZIP14, the most abundant ZIP in the liver, is upregulated by Fe deficiency( Reference Nam and Knutson 19 ). On the other hand, the ZnT family comprises 10 members so far and contribute to Zn homoeostasis by exporting Zn out of the cell or sequestrating Zn into cellular compartments when Zn levels are low. While there is evidence for direct action of Fe on ZIP, which may lead to alterations in Zn metabolism, interactions between Fe and ZnT have not been identified.
Metabolic crossroads between Fe, Cu and Zn during pregnancy are less known. Maternal Fe deficiency in rat increases Cu levels in the maternal liver, maternal serum as well as the placenta( Reference Sherman and Moran 20 , Reference Sherman and Tissue 21 ) along with an increase in serum Cu and CP( Reference Gambling, Dunford and McArdle 22 ). In contrast, we showed that Fe deficiency decreased Cu levels in the fetal liver, without affecting the fetal expression of the main genes involved in Cu metabolism( Reference Gambling, Andersen and Czopek 23 , Reference Lenartowicz, Kennedy and Hayes 24 ). In humans, Fe supplementation has been shown to decrease plasma Zn levels during pregnancy and Zn absorption during lactation in humans( Reference Fung, Ritchie and Woodhouse 25 – Reference Fischer-Walker, Kordas and Stoltzfus 27 ). Whether Fe deficiency, as opposed to overload, affects Zn metabolism in the mother and fetus remains essentially unknown. In this study, we investigated the effect of maternal Fe deficiency on Zn status in pregnant rats and the offspring as well as the implication of major genes of Zn metabolism. We also examined the mechanisms through which Fe deficiency differentially affects Cu metabolism in the mother and fetus and tested the implication of most genes of Cu metabolism in the liver of the mother and fetus as well as in the placenta.
Methods
Animal procedures
Experiments were performed using female Rowett hooded rats from three different studies of identical design. The protocol and animal procedures have recently been described in detail( Reference Cottin, Gambling and Hayes 28 ) and were approved by the Rowett Institute Ethics Committees and conducted in accordance with the UK Animals (Scientific Procedures) Act 1986. Briefly, 54 weanling females were fed a control diet (AIN93, containing 860 mg/kg Zn and 170 mg/kg Cu) for two weeks and were then randomised into two groups. In all, 30 remained on the control diet (Fe=50 mg/kg) for four weeks until mating and 24 switched to an Fe-deficient (Fe=7·5 mg/kg) diet for the same duration. They were mated with males of the same strain and remained on the same diet until they were killed at day 21 of gestation. Dams were killed by stunning and cervical dislocation and the fetuses by decapitation. Animals were housed under 12-h light–12-h dark cycle with a constant temperature (22°C). No adverse effects were recorded as a result of this dietary intervention.
Livers from the dams and fetuses, as well as the placentas, were collected and immediately frozen in liquid N2. They were kept in metal-free vials and stored at –80°C until analysis. Whenever possible, placentas and fetal livers from the three male median weights in each litter were pooled together and ground in liquid N2. Ground tissues from maternal liver, pooled placentas and pooled fetal livers were used for Fe, Cu and Zn content (control n 24, Fe deficient n 30) and gene analysis (control n 21, Fe deficient n 24). It should be noted that control and Fe-deficient samples from each of the three animal studies were analysed together, and results from each experiment were then pooled together for data analysis.
Assessment of copper and zinc status
Total Cu and Zn were measured in maternal liver, placenta and fetal liver by inductively coupled plasma (ICP)-MS as described previously( Reference Cottin, Gambling and Hayes 28 ). Samples were digested in 2 % HNO3, 0·5 % HCl and metal concentrations measured using an Agilent 7700X spectrophotometer (Agilent Technologies) equipped with a MicroMist nebuliser, Ni sampler and skimmer cones. Intra-assay CV% were 10·8 % (Fe), 11·1 % (Cu) and 5·2 % (Zn).
RNA extraction and real-time RT-quantitative PCR
In all, 20–30 mg of frozen placenta, maternal and fetal liver ground tissue were homogenised after grinding, using a T25 Ultra-Turrax (IKA (England) Ltd). RNA was isolated with the RNeasy Mini Kit (Qiagen), and 200 ng was reverse transcribed on a PTC-100 Thermal Cycler (Bio-Rad Laboratories) using the Applied Biosystems TaqMan RT reagent kit (Life Technologies). Complementary DNA fragments were amplified by real-time quantitative PCR (7700 Sequence Detection System; Life Technologies) using the primers (TaqMan; ThermoFisher Scientific) described in Table 1. The most relevant genes of Cu( Reference Lenartowicz, Kennedy and Hayes 24 ) and Zn metabolism were selected. Specifically, ZIP-1 is ubiquitously expressed and was shown to bind Fe with high affinity( Reference Segawa, Shibamoto and Ogawa 29 ). ZIP-4 is crucial during development and for the absorption of maternal Zn( Reference Andrews 30 ). ZIP8 and ZIP14 are closely related and have been shown to mediate Fe transport in addition to Zn( Reference Liuzzi, Aydemir and Nam 31 , Reference Wang, Jenkitkasemwong and Duarte 32 ). ZnT1 (ubiquitous), ZnT4 and ZnT5 are sensitive to maternal Zn status and may play an important role in fetal Zn supply from the maternal diet( Reference Helston, Phillips and McKay 33 , Reference Liuzzi, Bobo and Cui 34 ). The gene expression was normalised to 18s rRNA (HS99999901_s1) in all samples. This was chosen although the levels of expression were higher than the test genes because it was the only gene that was consistently unaffected by the nutritional treatments (G Roussel and HJ McArdle, unpublished results).
Table 1 List of genes analysed by PCR using TaqMan® Gene Expression Assays (ThermoFisher Scientific)
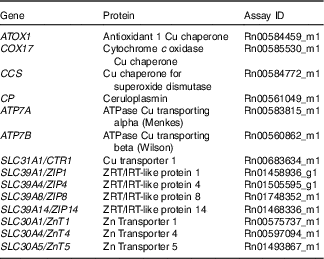
The experiment was performed and data interpreted according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines( Reference Bustin, Benes and Garson 35 ): 18s RNA expression did not significantly vary between the Fe-deficient and control groups in all tissues (Mann–Whitney test, data not shown). Linear amplification was observed for 18s and all genes of interest across their range of expression.
Statistical analysis
Fe, Cu and Zn content are expressed in µg/g dry tissue. The material used in this article were obtained from three separate experiments. To minimise variations between the three animal studies, as well as between each PCR run, the expression of Cu and Zn genes was expressed as percentage of the average gene expression of the control group for each of the three experiments. The normality of the distribution and equality of variances were analysed with GraphPad Prism (version 5.04). Data were analysed with IBM SPSS statistics 21.0 software using unpaired t tests when data were normally distributed and otherwise using non-parametric tests (Mann–Whitney U).
Results
Copper and zinc levels
The effect of maternal Fe deficiency on growth, placental:fetal ratio, placenta and liver weights, haematocrit and Fe levels in this study has been recently reported( Reference Cottin, Gambling and Hayes 28 ). Specifically, Fe levels were decreased by 152·6 µg/g (approximately –30 %), 604·0 µg/g (approximately –60 %) and 369·4 µg/g dry tissue (approximately –75 %), in the placenta, fetal and maternal liver, respectively( Reference Cottin, Gambling and Hayes 28 ). Maternal liver and placenta weight were not affected by Fe deficiency, while fetal weight and fetal liver weight were significantly decreased by approximately 13 % and 25 %, respectively( Reference Cottin, Gambling and Hayes 28 ). Maternal Fe deficiency increased Cu levels by 1·44 µg/g dry tissue (95 % CI 0·34, 2·54; P=0·002) and 3·45 µg/g (95 % CI –0·42, 7·32; P=0·018) in maternal liver and placenta, respectively. In contrast, Fe deficiency decreased Cu levels in the fetal liver by 9·64 µg/g (95 % CI –15·85, –3·42; P=0·006) (Fig. 1(a), Mann–Whitney), while Zn levels were increased by 81·6 µg/g (95 % CI 50·0, 113·1; P<0·0001). Zn levels in maternal liver and placenta were not significantly altered by maternal Fe deficiency (Fig. 1(b), t test). Changes in absolute levels of Fe, Zn and Cu followed a similar pattern (Table 2) apart from the absolute Zn levels that were reduced by 4·08 µg (95 % CI –1·64, –6·52; P=0·003) in the fetal liver.
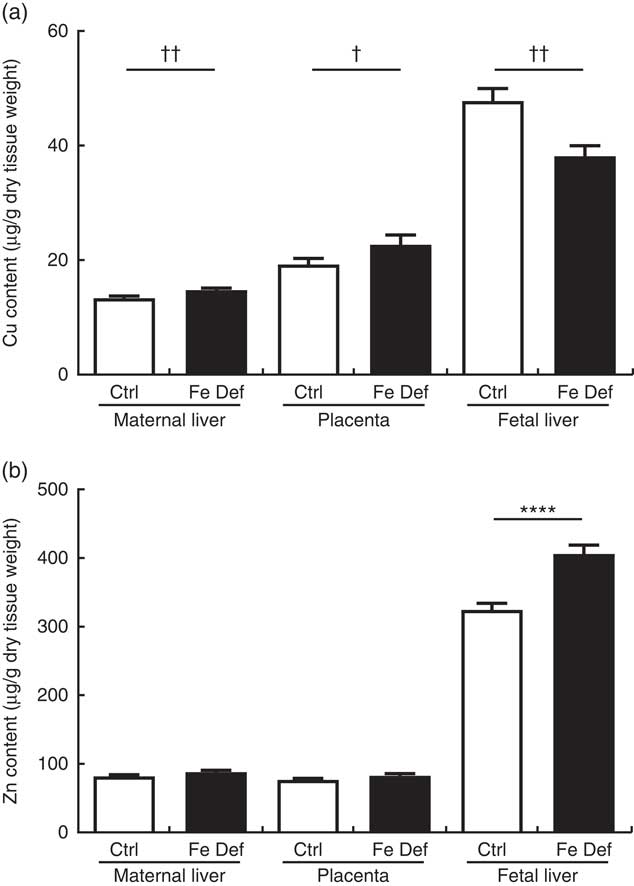
Fig. 1 Effect of maternal iron deficiency on copper (a) and zinc (b) levels in maternal liver, placenta and fetal liver 21 d after mating (n 54). Values are mean percentage of control, with their standard errors represented by vertical bars. Results are significantly different between the control (Ctrl, n 24) and iron-deficient (Fe Def, n 30) groups: **** P<0·0001 (independent t test); † P<0·05, †† P<0·01 (Mann–Whitney test).
Table 2 Organ weight, water content and absolute iron, copper and zinc content in the maternal liver, placenta and fetal liver of control and iron-deficient rats at day 21 of gestation (Mean values with their standard errors)
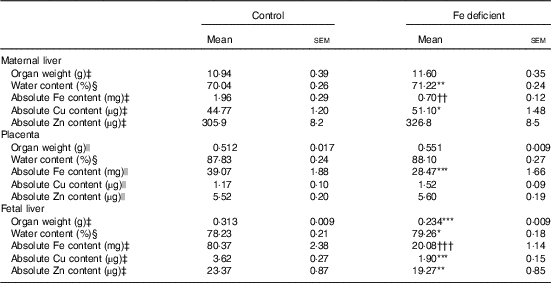
Mean values were significantly different compared with control:
* P<0·05
** P<0·01
*** P<0·001 (independent t test).
Mean values were significantly different compared with control:
†† P<0·01
††† P<0·001 (Mann–Whitney U test).
‡ n 16 (eight control, eight Fe deficient).
§ n 54 (twenty-four control, thirty Fe deficient).
|| n 38.
Zinc and copper gene expression
Maternal Fe deficiency decreased the hepatic expression of the Cu chaperones antioxidant 1 Cu chaperone (ATOX1) by –10·5 % (95 % CI –20·7, –0·4; P=0·042) and cytochrome c oxidase Cu chaperone (COX17) by –13·8 % (95 % CI –25·3, –2·3; P=0·020) in the dams. In contrast, the hepatic expression of COX17 was increased by 15·0 % (95 % CI 2·5, 27·5; P=0·020) in the fetus. In the placenta, Fe deficiency led to a decrease in Cu chaperone for superoxide dismutase (CCS), the chaperone that delivers Cu to superoxide dismutase (SOD)1, by –9·5 % (95 % CI –18·0, –1·0; P=0·030) and CP by –15·0 % (95 % CI –29·4, –0·5; P=0·042) expression (Fig. 2).
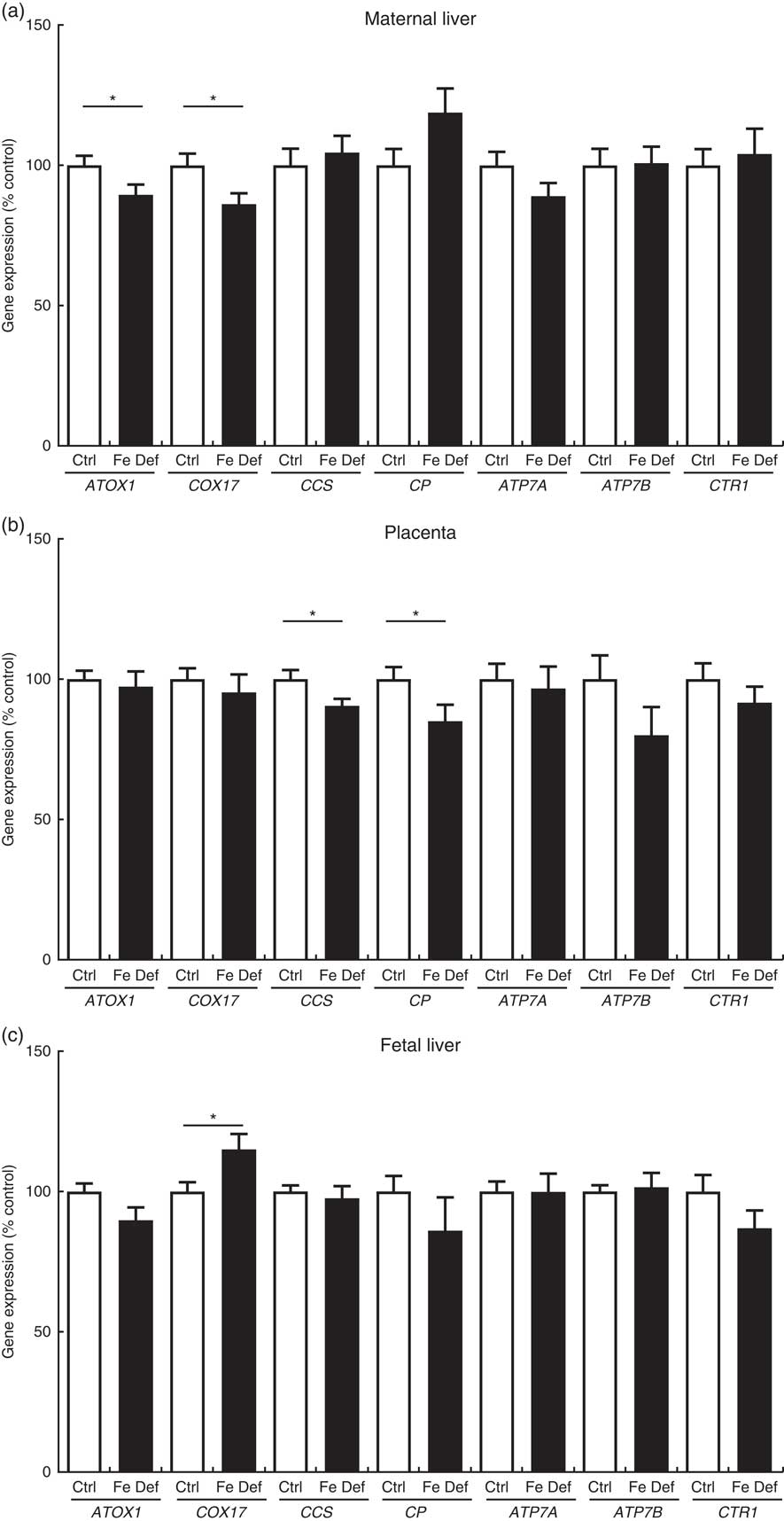
Fig. 2 Effect of maternal iron deficiency on the expression of genes related to copper metabolism (n 45) 21 d after mating: (a) maternal liver; (b) placenta; (c) fetal liver. Values are mean percentage of control, with their standard errors represented by vertical bars. * Results are significantly different between the control (Ctrl, n 21) and iron-deficient (Fe Def, n 24) groups (P<0·05; Mann–Whitney test). ATOX1, antioxidant 1 copper chaperone; COX17, cytochrome c oxidase chaperone; ATP7A, ATPase copper transporting alpha (Menkes); ATP7B, ATPase copper transporting beta (Wilson); CCS, copper chaperone for superoxide dismutase; CP, ceruloplasmin; CTR1, copper transporter 1.
The maternal hepatic expression of the genes related to Zn metabolism was not affected by Fe deficiency. In the placenta, ZIP4 was reduced by –23·7 % (95 % CI –47·0, –0·4; P=0·047) and ZnT1 by –12·3 % (95 % CI 21·8, –2·8; P=0·012) in the Fe-deficient group compared with control (Fig. 3). In fetal liver, Fe deficiency also decreased ZIP4 by –17·0 % (95 % CI –28·1, –5·8; P=0·004), while ZIP14 and ZnT1 were increased by 10·0 % (95 % CI 0·7, 19·3; P=0·036) and by 18·7 % (95 % CI 9·0, 28·4; P=0·0003), respectively (Fig. 3).
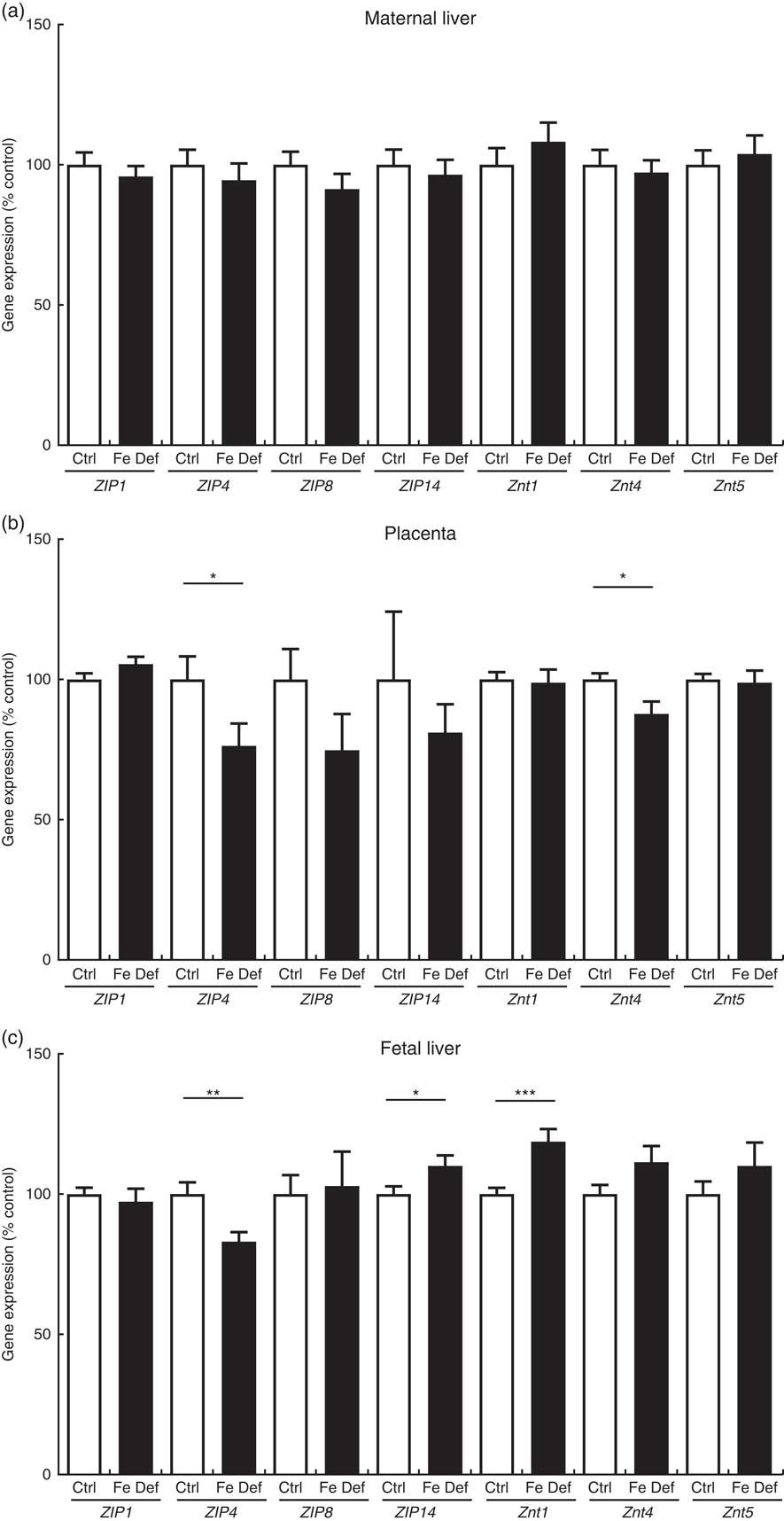
Fig. 3 Effect of maternal iron deficiency on the expression of genes related to zinc metabolism (n 45) 21 d after mating: (a) maternal liver; (b) placenta; (c) fetal liver. Values are mean percentage of control, with their standard errors represented by vertical bars. Results are significantly different between the control (Ctrl, n 21) and iron-deficient (Fe Def, n 24) groups: * P<0·05, ** P<0·01, *** P<0·001 (Mann–Whitney test). ZIP1, ZRT/IRT-like protein 1; ZIP4, ZRT/IRT-like protein 4; ZIP8, ZRT/IRT-like protein; ZIP14, ZRT/IRT-like protein 14; ZnT1, zinc transporter 1; ZnT4, zinc transporter 4; ZnT5, zinc transporter 5.
The effect of maternal Fe deficiency on Cu and Zn metabolism observed in the current study is summarised in Table 3.
Table 3 Summary of the effect of maternal iron deficiency on copper and zinc metabolism in maternal liver, placenta and fetal liver at day 21 of pregnancy *
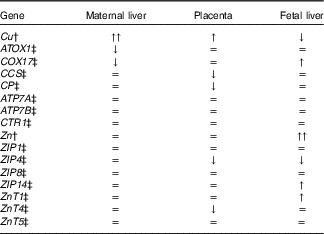
ATOX1, antioxidant 1 Cu chaperone; COX17, cytochrome c oxidase Cu chaperone; CCS, Cu chaperone for superoxide dismutase; CP, ceruloplasmin; ATP7A, ATPase Cu transporting alpha (Menkes); ATP7B, ATPase Cu transporting beta (Wilson); CTR1, Cu transporter 1; ZIP, ZRT/IRT-like protein; ZnT, Zn transporter.
* The concentrations of Cu, Zn, and the expression of genes of Cu and Zn metabolism were either greatly increased (↑↑), increased (↑), decreased (↓) or unchanged (=) by maternal Fe deficiency.
† n 54 (control n 24, Fe deficient n 30).
‡ n 45 (control n 21, Fe deficient n 24).
Discussion
In this article, we examined the interactions between Fe, Cu and Zn during pregnancy in the rat. The study has some limitations in that the data can only be extrapolated to humans to a limited extent. However, given how well-conserved micronutrient metabolism is between species, it is likely that the same results would be observed in humans with Fe deficiency. We restricted our measurements to liver and placenta, as these are the major organs involved in micronutrient metabolism( Reference Gambling, Kennedy and McArdle 3 ). Fe deficiency altered Cu but not Zn concentrations in the maternal liver and placenta, confirming that the metabolism of Cu is more sensitive to Fe deficiency than that of Zn( Reference Stangl and Kirchgessner 15 ). However, maternal Fe deficiency had an important impact on both Cu and Zn levels in the fetal liver, which were reduced by approximately 15 % and augmented by approximately 25 %, respectively.
Effect of maternal iron deficiency on copper metabolism
Maternal Fe deficiency leads to the hepatic accumulation of Cu in the dams, while Cu levels are decreased in the fetal liver, as described previously by our group, as well as others( Reference Sherman and Moran 20 , Reference Sherman and Tissue 21 , Reference Gambling, Andersen and Czopek 23 ). Interestingly, Fe deficiency decreased the expression of the chaperones ATOX1 and COX17 in the maternal liver, contrasting with our previous findings, where no change in ATOX1 mRNA levels was observed( Reference Gambling, Andersen and Czopek 23 ). It is important to note that in our previous experiment, COX17 was not measured and ATOX1 was only measured in six samples. Further, the previous study used Northern blotting to detect expression levels which is less sensitive than using RT-PCR. The present study included 45 samples, which allows us to detect smaller changes with greater sensitivity.
ATOX1 does not seem to be involved in Cu uptake but is a Cu chaperone that interacts with ATP7A and ATP7B and delivers Cu to these pumps and hence to trans-Golgi network( Reference Kambe, Weaver and Andrews 36 ) or possibly the efflux pathway of the placenta( Reference Hardman, Michalczyk and Greenough 37 ). Moreover, ATOX1 appears essential to peri-natal Cu homoeostasis( Reference Hamza, Faisst and Prohaska 38 ) for synthesis of CP or export from the cell into the portal circulation (ATP7A in gut) or into the bile for excretion (ATP7B). Understandably, the inhibition of ATOX1 expression reduces the trafficking of ATPases( Reference Hamza, Prohaska and Gitlin 39 ), thus reducing Cu export and causing its accumulation in the cell( Reference Safaei, Maktabi and Blair 40 ). Therefore, the small but significant reduction in ATOX1 expression in the Fe-deficient liver could be responsible – at least partially – for the increase in Cu levels.
In addition, the hepatic expression of COX17, a chaperone delivering Cu to the cytochrome oxidase complex in the respiratory chain, was also decreased. The impairment of mitochondrial function and the reduction in cytochrome c activity (which requires both Fe and Cu to function) is well-established in Fe-deficient tissues, including in the liver( Reference Masini, Salvioli and Cremonesi 41 , Reference Walter, Knutson and Paler-Martinez 42 ), although whether this is cause or consequence is not certain at this time. There are little data on the direct effects of Cu on either ATOX1 or COX17 expression, so the possibility that these changes are a response to changes in Cu levels, rather than Cu levels being changed by the alterations in gene expression, cannot be excluded in these experiments.
As observed previously( Reference Gambling, Andersen and Czopek 23 ), maternal Fe deficiency led to the increase in Cu levels in the placenta (approximately 19 %) to a greater extent than the maternal liver (approximately 10 %). This was accompanied by the reduction in CCS expression, another chaperone channeling Cu for incorporation into the Cu–Zn superoxide dismutase (SOD) complex, an essential component of the anti-oxidant cell system. Cu has been shown to modulate the degradation of CCS ( Reference Bertinato and L’Abbé 43 ) and Cu supplementation in humans reduces CCS mRNA levels in peripheral mononuclear cells( Reference Araya, Andrews and Pizarro 44 ). It is likely that the down-regulation of CCS expression observed here is due to the presence of elevated Cu levels in the placenta, rather than a direct consequence of reduced Fe levels. Our findings suggest that maternal Fe deficiency leads to the impairment of SOD activity (closely related to CCS expression) and the anti-oxidant defense of the placenta as observed in liver and brain cells upon Cu accumulation( Reference Ozcelik and Uzun 45 , Reference Zhang, Noordin and Rahman 46 ).
CP is involved in Fe export in the liver, oxidising Fe(ii) to Fe(iii) for incorporation into transferrin, while its homologue zyklopen performs the same function in placenta in mice and humans( Reference Chen, Attieh and Syed 47 ). Whether the ferroxidase mRNA measured in the placenta is zyklopen, CP or a combination of both remains uncertain, since the sequences are very similar and because the primers are from a commercial supplier. However, it is likely that we measured the mRNA coding for a multi-copper oxidase responsible for Fe export( Reference Danzeisen, Fosset and Chariana 48 ). The effect that the reduced placental expression might have on Fe transport is not clear and contrasts with previous findings( Reference Danzeisen, Fosset and Chariana 48 ). However, it is important to note that CP protein and enzyme levels do not correlate with mRNA levels( Reference McArdle, Mercer and Sargeson 49 ) and that changes in CP mRNA expression observed in the present study may therefore have little physiological impact. Maternal Fe deficiency is partly alleviated in the fetus, due to increased expression of transferrin receptors( Reference Cornock, Gambling and Langley-Evans 50 , Reference Garcia-Valdes, Campoy and Hayes 51 ), which would imply that ferroxidase expression or activity is not rate limiting.
Cu transport across the placenta is less well-understood. Cu appears to be taken up from the maternal circulation through CTR1 and excreted through ATPases( Reference Kuo, Zhou and Cosco 52 , Reference McArdle, Andersen and Jones 53 ). It has been suggested that ATP7A is responsible for the efflux of Cu from the basolateral membrane into the fetal circulation, while ATP7B is responsible for excreting Cu back into the maternal circulation at the apical side( Reference Hardman, Manuelpillai and Wallace 54 ). Whether CP and/or zyklopen are involved in Cu efflux and/or delivery to the fetal liver, as we have previously( Reference Hardman, Manuelpillai and Wallace 54 ) suggested for transport of Cu from the mother to the placenta( Reference Hilton, Spenser and Ross 55 ), and whether this could explain the differences observed in Cu levels between placenta and fetal liver remain to be clarified.
Effects of maternal iron deficiency on zinc metabolism
The effect of Fe deficiency on Zn metabolism in the pregnant rat is less marked than for Cu but is nonetheless significant. Zn levels in the maternal liver and placenta were not changed but were markedly increased in the fetal liver.
In the maternal liver, the expression of the genes of Zn metabolism tested were unchanged, which is consistent with the lack of effect on Zn levels but contrasts with previous findings in male rats, where Fe deficiency led to an increase in hepatic ZIP14 expression( Reference Nam and Knutson 19 ). It should be noted, however, that the authors also observed a small decrease in Zn levels which was not seen in the present study nor in their other study of a similar design( Reference Nam and Knutson 19 ). Taken together, these results imply that Fe deficiency during pregnancy is unlikely to lead to changes in Zn metabolism that are physiologically important in the maternal liver.
While Zn levels were unchanged in the placenta, ZIP4 and ZnT4, genes of Zn cellular uptake and exit, respectively, were both decreased in the placenta. The results do not fully match those seen in mice given a Zn-deficient diet, where ZnT4 and also ZIP1, ZnT1 and 5 were reduced in placenta( Reference Helston, Phillips and McKay 33 ). It is possible that there was a mild reduction in Zn supply to and in efflux from the placenta, with a neutral net effect on Zn levels in the placenta. Zn metabolism and transport in the placenta are still not entirely understood, and the role and localisation of ZIP4 in the placenta are not known. ZIP4 is crucial for Zn cellular uptake in the small intestine and is up-regulated upon Zn deficiency in mice( Reference Dufner-Beattie, Wang and Kuo 56 ). It is also abundantly expressed in other tissues involved in nutrient absorption and reabsorption (stomach, colon and kidney)( Reference Wang, Zhou and Kuo 57 ). While ZIP4 expression could not be compared with intestinal or kidney expression in the present study, it is important to note that it was higher in the placenta compared with maternal liver (approximately 2-fold) and fetal liver (approximately 15-fold, data not shown). This suggests that the changes observed in placental expression could be biologically relevant, and further research is warranted to clarify the role of ZIP4 in placenta. If it was involved in the return of Zn from fetal to maternal circulation, then decreased expression could result in increased levels of Zn in the fetal liver. Interestingly, neither ZIP8 nor ZIP14, both of which have previously been implicated in Fe metabolism as well as that of Zn( Reference Wang, Jenkitkasemwong and Duarte 32 ), are altered in the placenta or maternal liver, which would argue against the changes in fetal liver Zn levels being a direct consequence of competition with Fe for these transporters.
Fe deficiency leads to an increase in Zn levels in the fetal liver but also a lesser decrease in Fe levels compared with the decrease observed in the maternal liver( Reference Gambling, Charania and Hannah 58 ). Both these changes may be explained, at least partially, by an upregulation of ZIP14, which mediates the uptake of Zn, as well as non-transferrin-bound Fe( Reference Liuzzi, Aydemir and Nam 31 ), and thus could help protect the fetus against Fe deficiency. It is important to note that this does not rule out other mechanisms, such as an increased expression of transferrin receptors, as we have previously observed in the placenta of rats and women( Reference Cornock, Gambling and Langley-Evans 50 , Reference Garcia-Valdes, Campoy and Hayes 51 ). On the other hand, the reduced ZIP4 expression in the fetal liver is more likely to be a consequence of the increase in Zn levels as has been seen in Zn overload observed in the intestine and visceral yolk sac of mice( Reference Dufner-Beattie, Wang and Kuo 56 ). In addition, as described earlier in this discussion, ZIP4 expression in the fetal liver was fairly low compared with the other tissues, indicating that its changes may have little biological relevance.
Contrasting with our results, ZnT1 hepatic upregulation has been reported in Zn-deficient rats in association with Zn depletion in the liver( Reference Jou, Hall and Philipps 59 ), although this was only observed at the protein level. Whether the increase in ZnT1 mRNA expression in the fetal liver would lead to an increase in protein levels is uncertain, and whether this could lead to more Zn efflux back into the fetal circulation is unlikely in the present study. The reasons why ZnT1 upregulation is associated with a marked increase in Zn levels are not known, and the role of ZnT1 in fetal liver Zn metabolism needs to be clarified.
In summary, this study has shown that Fe deficiency has opposite effects on Cu and Zn levels in the fetal liver, and this is associated with changes in the expression of genes of Cu and Zn metabolism in the placenta as well as in the fetus. It is important to note that some of these effects may not be directly caused by Fe deficiency but rather by changes in the metabolism of the other two nutrients. The results further demonstrate that micronutrient metabolism, especially during pregnancy, is tightly interlinked and underscores the importance of considering all of the micronutrients when trying to alleviate deficiencies in one of them. It would be important to consider that the symptoms caused by deficiencies in one micronutrient (here Fe) may not be caused directly by that deficiency but by other deficiencies or overloads occurring as a consequence of the primary defect.
Acknowledgements
The authors are grateful for the technical assistance of Gill Campbell (ICP-MS analysis).
This work was supported and funded by Scottish Government (Rural and Environment Science and Analytical Services).
H. J. M., L. G. and S. C. C. designed the research; L. G., H. E. H., V. J. C. and S. C. C. conducted the research; S. C. C. analysed the data and wrote the manuscript; G. R. and H. J. M. reviewed the manuscript. H. J. M., G. R. and S. C. C. take responsibility of data interpretation and presentation. All authors read and approved the final manuscript.
The authors declare that there are no conflicts of interest.






