Studies of maternal mental health (O'Donnell, Glover, Barker, & O'Connor, Reference O'Donnell, Glover, Barker and O'Connor2014; O'Donnell & Meaney, Reference O'Donnell and Meaney2016; Pawlby, Hay, Sharp, Waters, & O'Keane, Reference Pawlby, Hay, Sharp, Waters and O'Keane2009), institutional rearing (McLaughlin et al., Reference McLaughlin, Sheridan, Winter, Fox, Zeanah and Nelson2014; Nelson et al., Reference Nelson, Zeanah, Fox, Marshall, Smyke and Guthrie2007; Rutter, Kumsta, Schlotz, & Sonuga-Barke, Reference Rutter, Kumsta, Schlotz and Sonuga-Barke2012; Sonuga-Barke et al., Reference Sonuga-Barke, Kennedy, Kumsta, Knights, Golm, Rutter and Kreppner2017), and childhood abuse (Klengel et al., Reference Klengel, Mehta, Anacker, Rex-Haffner, Pruessner, Pariante and Binder2013; Nanni, Uher, & Danese, Reference Nanni, Uher and Danese2012; Spatz Widom, DuMont, & Czaja, Reference Spatz Widom, DuMont and Czaja2007) reflect the profound influence of the early environment on child development and later mental health. However, not all children are affected equally, with a subset of children seemingly resilient to some of the negative consequences of early life adversity. Early intervention programs that seek to mitigate the negative sequelae associated with early life adversity would benefit from a better understanding of such resilience.
The genome is one source of individual differences in sensitivity to the early environment. The negative effects of maternal antenatal anxiety on child emotional problems (O'Donnell, Glover, Holbrook, & O'Connor, Reference O'Donnell, Glover, Holbrook and O'Connor2014), working memory, and symptoms of ADHD (O'Donnell et al., Reference O'Donnell, Glover, Lahti, Lahti, Edgar, Raikkonen and O'Connor2017) are moderated, in part, by genetic variation in the child. Likewise, polygenic variation across the serotonin and dopamine signaling pathways moderates the impact of prenatal adversity (Silveira et al., Reference Silveira, Pokhvisneva, Parent, Cai, Rema, Broekman and Meaney2017) and early caregiving (Belsky & van IJzendoorn, Reference Belsky and van IJzendoorn2017; Stocker et al., Reference Stocker, Masarik, Widaman, Reeb, Boardman, Smolen and Conger2017) on child neurodevelopment and long-term mental health outcomes.
The quality of the early caregiving environment can also contribute to individual differences in susceptibility to the negative effects of early adversity. Bergman, Sarkar, Glover, and O'Connor (Reference Bergman, Sarkar, Glover and O'Connor2010) highlight infant attachment security as a moderator of prenatal adversity effects on infant cognitive development. In their study of women undergoing amniocentesis, the authors report an inverse association between fetal cortisol exposure and measures of cognitive development at 17 months. This relationship was only evident in infants with an insecure attachment style. Likewise, McGoron et al. (Reference McGoron, Gleason, Smyke, Drury, Nelson, Gregas and Zeanah2012) report that infant attachment security limited the impact of institutional rearing on measures of child psychopathology. Taken together, these findings suggest the early caregiving environment can buffer some of the negative consequences of adversity experienced in the prenatal and early postnatal environment. The biological basis of such effects is not well understood.
Chemically stable, epigenetic modifications, such as DNA methylation, have emerged as a putative mechanism for the enduring effects of the early environment on neural development and function (Meaney & Ferguson-Smith, Reference Meaney and Ferguson-Smith2010). DNA methylation can modulate gene expression as well as splicing events (Maunakea, Chepelev, Cui, & Zhao, Reference Maunakea, Chepelev, Cui and Zhao2013; Maunakea et al., Reference Maunakea, Nagarajan, Bilenky, Ballinger, D'Souza, Fouse and Costello2010), thus creating a diversity of transcriptional programs from a single genome. The human DNA methylome is dynamic across development (Teh et al., Reference Teh, Pan, Chen, Ong, Dogra, Wong and Holbrook2014; Ziller et al., Reference Ziller, Gu, Muller, Donaghey, Tsai, Kohlbacher and Meissner2013) and is altered by early social experience (Klengel et al., Reference Klengel, Mehta, Anacker, Rex-Haffner, Pruessner, Pariante and Binder2013; Lam et al., Reference Lam, Emberly, Fraser, Neumann, Chen, Miller and Kobor2012; O'Donnell et al., Reference O'Donnell, Chen, MacIsaac, McEwen, Nguyen, Beckmann and Meaney2018). Studies using rodent models reveal effects of parent–offspring interactions on the methylation status of a range of genes implicated in hypothalamic–pituitary–adrenal stress responses as well as synaptic function (Bagot et al., Reference Bagot, Zhang, Wen, Nguyen, Nguyen, Diorio and Meaney2012; Klengel et al., Reference Klengel, Mehta, Anacker, Rex-Haffner, Pruessner, Pariante and Binder2013; Lam et al., Reference Lam, Emberly, Fraser, Neumann, Chen, Miller and Kobor2012; Murgatroyd et al., Reference Murgatroyd, Patchev, Wu, Micale, Bockmuhl, Fischer and Spengler2009; O'Donnell et al., Reference O'Donnell, Chen, MacIsaac, McEwen, Nguyen, Beckmann and Meaney2018; Roth, Lubin, Funk, & Sweatt, Reference Roth, Lubin, Funk and Sweatt2009; Weaver et al., Reference Weaver, Cervoni, Champagne, D'Alessio, Sharma, Seckl and Meaney2004; Wu, Patchev, Daniel, Almeida, & Spengler, Reference Wu, Patchev, Daniel, Almeida and Spengler2014; Zhang et al., Reference Zhang, Hellstrom, Bagot, Wen, Diorio and Meaney2010).
Genome-wide analyses of DNA methylation in humans reveal broad effects of childhood abuse (Mehta et al., Reference Mehta, Klengel, Conneely, Smith, Altmann, Pace and Binder2013; O'Donnell et al., Reference O'Donnell, Chen, MacIsaac, McEwen, Nguyen, Beckmann and Meaney2018; Suderman et al., Reference Suderman, McGowan, Sasaki, Huang, Hallett, Meaney and Szyf2012). However, few studies examine whether variations in parent–offspring interactions lying within the normal range are a source of variation in the DNA methylome. One recent study reports an association between one specific component of caregiving, the amount of physical contact between mothers and their infants, and variation in DNA methylation (Moore et al., Reference Moore, McEwen, Quirt, Morin, Mah, Barr and Kobor2017). However, Moore et al. did not examine how the quality of such mother–infant interactions predicted variation in the DNA methylome.
Infant attachment is an objective measure of the early care environment that associates with child mental health outcomes (Fearon, Bakermans-Kranenburg, van IJzendoorn, Lapsley, & Roisman, Reference Fearon, Bakermans-Kranenburg, van IJzendoorn, Lapsley and Roisman2010; Groh et al., Reference Groh, Fearon, Bakermans-Kranenburg, van IJzendoorn, Steele and Roisman2014; Groh, Roisman, van IJzendoorn, Bakermans-Kranenburg, & Fearon, Reference Groh, Roisman, van Ijzendoorn, Bakermans-Kranenburg and Fearon2012) and may buffer the effects of prenatal adversity on measures of child development (Bergman et al., Reference Bergman, Sarkar, Glover and O'Connor2010; McGoron et al., Reference McGoron, Gleason, Smyke, Drury, Nelson, Gregas and Zeanah2012). In this study we use a prospective, longitudinal cohort of typically developing children to describe the association between infant attachment and variation in DNA methylation across the genome.
Method and Materials
Participants
Participants (N = 226: 111 female, 115 male, mean age = 6.99, SD = 1.33 years) were drawn from the Maternal Adversity, Vulnerability and Neurodevelopment (MAVAN) cohort (O'Donnell, Gaudreau, et al., Reference O'Donnell, Gaudreau, Colalillo, Steiner, Atkinson and Moss2014). Full ethical approval for this study was provided by the institutional review boards at the Douglas Research Institute and McMaster University.
The early care environment
Infant attachment was assessed at 36 months using a modified version of the Strange Situation (Moss, Bureau, Cyr, Mongeau, & St.-Laurent, Reference Moss, Bureau, Cyr, Mongeau and St.-Laurent2004). This 20-min observational assessment consists of a sequence of separations/reunions lasting 5 min each: (1) separation between mother and child; (2) reunion; (3) second separation; and (4) second reunion. Behavior was coded during each reunion episode giving rise to one of four attachment classifications: secure, avoidant, ambivalent, or disorganized (Moss, Cyr, Bureau, Tarabulsy, & Dubois-Comtois, Reference Moss, Cyr, Bureau, Tarabulsy and Dubois-Comtois2005). A secure attachment style is characterized by increased exploration in the presence of the primary caregiver and generally associates with more favorable developmental outcomes. Insecure attachment is characterized by reduced exploration, even in the presence of the primary caregiver. Insecurely attached infants are further subdivided as either “avoidant” of the primary caregiver during stress or striving to maintain close proximity to the primary caregiver, but not soothed during distress (ambivalent). Disorganized attachment describes infants who fail to show an “organized” strategy for eliciting the attention of the primary caregiver during stress, who display stress-associated behaviors (e.g., hunched shoulders or freezing) and may show evidence of role-reversed behavior (e.g., attempting to control the caregiver; Moss et al., Reference Moss, Cyr, Bureau, Tarabulsy and Dubois-Comtois2005). Disorganized attachment is also associated with an increased risk of later psychopathology (Fearon et al., Reference Fearon, Bakermans-Kranenburg, van IJzendoorn, Lapsley and Roisman2010; Groh et al., Reference Groh, Roisman, van Ijzendoorn, Bakermans-Kranenburg and Fearon2012; Scholtens, Rydell, Bohlin, & Thorell, Reference Scholtens, Rydell, Bohlin and Thorell2014). Trained observers scored all videos and demonstrated excellent interrater reliability (κ = 0.83). Due to the small number of avoidant (n = 13) and ambivalent (n = 25) cases, both groups were combined to form an organized–insecure category.
Biological sample collection
We collected buccal epithelial cells (Catch-All Swabs, Epicentre, USA) from all children (mean age = 6.99, SD = 1.33 years). Buccal cell collection is minimally invasive, provides a relatively homogenous cell type (squamous epithelium) for DNA methylation analyses (Berko et al., Reference Berko, Suzuki, Beren, Lemetre, Alaimo, Calder and Greally2014; Lowe et al., Reference Lowe, Gemma, Beyan, Hawa, Bazeos, Leslie and Ramagopalan2013), and may serve as an appropriate surrogate for neural tissue given their shared ectodermal lineage. Papavassiliou et al. (Reference Papavassiliou, York, Gursoy, Hill, Nicely, Sundaram and Jackson-Cook2009) report that trisomy mosaicism in buccal cells is a stronger predictor of cognitive impairment in Down syndrome patients than mosaicism determined in lymphocytes (Papavassiliou et al., Reference Papavassiliou, York, Gursoy, Hill, Nicely, Sundaram and Jackson-Cook2009). Likewise, Smith et al. (Reference Smith, Kilaru, Klengel, Mercer, Bradley, Conneely and Binder2015) report greater similarity between DNA methylome profiles derived from neural tissue and saliva than blood-based data. Smith et al. suggest that this brain-saliva similarity is driven, in part, by the proportion of epithelial cells within saliva samples.
DNA methylation analyses
Genomic DNA was extracted using the Masterpure system (Epicentre, USA) and quantified using a NanoPhotometer P300 (Implen, Germany). The genomic DNA (750 ng) was bisulfite converted using the EZ-DNA Methylation Kit (Zymo Research, USA) and isothermally amplified at 37 °C for 22 hr, enzymatically fragmented, purified, and hybridized on Infinium HumanMethylation450 beadchip array (450K, Illumina). The 450K array permits quantification of DNA methylation with single-base resolution across multiple probes, including 482,421 CpG sites and 3,091 non-CpG sites (Bibikova et al., Reference Bibikova, Barnes, Tsan, Ho, Klotzle, Le and Shen2011). Our studies with this technology suggest strong reproducibility and good correlation with bisulfite sequencing, including reduced representational bisulfite sequencing and confirmatory pyrosequencing (Marr et al., Reference Marr, MacIsaac, Jiang, Airo, Kobor and McMaster2014; O'Donnell et al., Reference O'Donnell, Chen, MacIsaac, McEwen, Nguyen, Beckmann and Meaney2018; Pan et al., Reference Pan, Chen, Dogra, Ling Teh, Tan, Lim and Holbrook2012). The 450K chips were scanned using the Illumina HiScan system and the image data were processed in R (R Core Team, 2014) using the Minfi package (Aryee et al., Reference Aryee, Jaffe, Corrada-Bravo, Ladd-Acosta, Feinberg, Hansen and Irizarry2014).
The experiment was run in 4 batches together with a series of internal controls distributed across each batch and longitudinal samples (Forest et al., Reference Forest, O'Donnell, Voisin, Gaudreau, MacIsaac, McEwen and Greenwood2018). We report findings from DNA methylation data generated from the first biological sample collection, which children provided at age 6.99 years (SD = 1.33).
450K data processing
DNA methylation data were processed in R using the Minfi package (Aryee et al., Reference Aryee, Jaffe, Corrada-Bravo, Ladd-Acosta, Feinberg, Hansen and Irizarry2014). Samples that failed standard Minfi quality control (QC; QC threshold = 10.5) were removed (Aryee et al., Reference Aryee, Jaffe, Corrada-Bravo, Ladd-Acosta, Feinberg, Hansen and Irizarry2014). All remaining samples had a high call rate (>95%). Probes with a low call rate (<75%), a high detection p value (p > .05; Lehne et al., Reference Lehne, Drong, Loh, Zhang, Scott, Tan and Chambers2015), and a low number of beads (less than 3 in >5% of the cohort) were also removed. Predicted sex (from DNA methylation of the sex chromosomes; Aryee et al., Reference Aryee, Jaffe, Corrada-Bravo, Ladd-Acosta, Feinberg, Hansen and Irizarry2014) and reported sex for all participants was consistent for all samples. Nonspecific probes or probes with single nucleotide polymorphism (SNP)-disrupting polymorphisms were removed (Chen et al., Reference Chen, Lemire, Choufani, Butcher, Grafodatskaya, Zanke and Weksberg2013; Price et al., Reference Price, Cotton, Lam, Farre, Emberly, Brown and Kobor2013), along with all the probes on the sex chromosomes. Unwanted technical variation was accounted for, in part, using functional normalization based on principal component analysis of control probes on the 450K (Fortin et al., Reference Fortin, Labbe, Lemire, Zanke, Hudson, Fertig and Hansen2014). We also used ComBat (Johnson, Li, & Rabinovic, Reference Johnson, Li and Rabinovic2007) to iteratively adjust our data for unwanted technical variation associated with experimental batch, array row (sentrix row), and plate position (sample column). Hierarchical clustering showed that technical replicates were more closely related to each other than to other individuals. Therefore, replicates from the same individual were combined such that each participant contributed one unique data point in our analyses. The data set was further reduced to probes showing interindividual variation, defined as probes with a range of methylation (β value ≥ 0.10) corresponding to at least a 10% difference in DNA methylation across participants. A total of 252,439 probes remained and were denoted as variably methylated probes (VMPs).
Cell type adjustments
We used the method described by Smith et al. (Reference Smith, Kilaru, Klengel, Mercer, Bradley, Conneely and Binder2015) to predict buccal epithelial cell content of each sample. The proportion of buccal cells per sample was added as a covariate to all models describing variation in DNA methylation.
Genetic analyses
We used the PsychChip and the commercially available PsychArray (Illumina) to describe genetic variation within this cohort. Only shared SNPs (between the PsychChip and the PsychArray) were considered. SNPs with a low call rate (i.e., called in <95% of samples) and samples with a low call rate (i.e., <95% of SNPs called within a sample) were removed, resulting in 223 participants and 228,562 autosomal SNPs. Next, we used the Sanger Imputation Service to impute missing genotypes, resulting in ~17M SNPs per participant.
Population structure
We filtered the genetic data set to retain SNPs in low linkage disequilibrium (r 2 < .25). These SNPs were then subjected to principal component analysis (PCA) in EIGENSTRAT, an established method to describe population structure (Price et al., Reference Price, Patterson, Plenge, Weinblatt, Shadick and Reich2006). Two principal component scores were included in all analyses to account for population stratification/ancestry effects.
Child outcome measures
We used the Bayley Scales of Infant Development II (BSID) to assess child motor development (Psychomotor Development Index; PDI) and mental development (Mental Development Index; MDI) at 36 months (Bayley, Reference Bayley1993). We also rated child behavior (Behavior Rating Scale; BRS) during the BSID across multiple domains (e.g., attention/arousal, orientation/engagement, emotional regulation, and motor quality) to index child performance during this mildly stressful, novel task.
Mothers completed a series of questionnaires concurrent with buccal sample collection from their children (see Table 1 for maternal and child age at time of collection). We used the maternal report from the Strengths and Difficulties Questionnaire (SDQ; Goodman, Meltzer, & Bailey, Reference Goodman, Meltzer and Bailey1998) to describe symptoms of child emotional/behavioral problems. This well-validated screening assessment provides symptom data on four domains related to emotional difficulties, inattention/hyperactivity, conduct problems, and peer relations. A summary score of these four problem subscales contributes to the total SDQ score, which shows good predictive validity for child mental health problems (Goodman, Ford, Simmons, Gatward, & Meltzer, Reference Goodman, Ford, Simmons, Gatward and Meltzer2000; Goodman, Renfrew, & Mullick, Reference Goodman, Renfrew and Mullick2000). Mothers also completed the Spielberger State/Trait Anxiety Inventory (Spielberger, Gorusch, & Lushene, Reference Spielberger, Gorusch and Lushene1970), which we used to index maternal trait anxiety at the time of biological sample collection from their child. Finally, maternal education (university educated or not) was coded as a binary variable and considered in all analyses.
Table 1. MAVAN 450K cohort characteristics
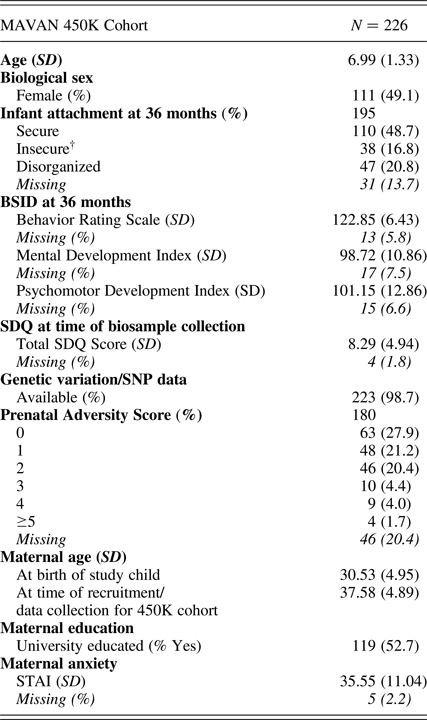
Note: BSID, Bayley Scales of Infant Development. SDQ, Strengths and Difficulties Questionnaire. STAI, Spielberger Trait Anxiety Inventory. †Avoidant (n = 13) and ambivalent (n = 25) attachment classifications were combined to form an “organized” insecure group.
Data analysis
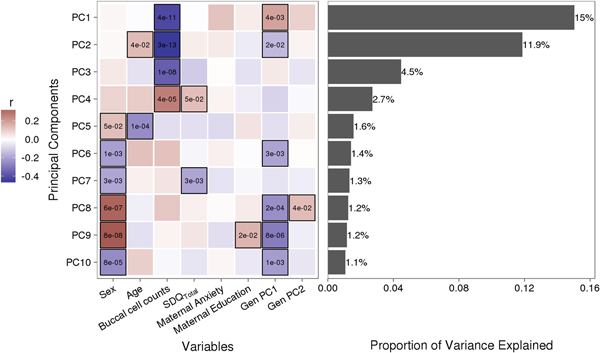
Variably methylated probes (VMPs; n = 252,439) were subjected to PCA in R. We focus our analysis on the first 10 principal components (PCs) that account for 41.9% of the variance in our DNA methylation data (Figure 1). We used one-way analysis of variance (ANOVA) models to describe associations between infant attachment and PCs scores. ANOVA models were adjusted for measures of cellular heterogeneity in buccal samples, child biological sex and age, measures of population stratification derived from genetic data, and maternal anxiety and level of education. Attachment style was coded as a categorical nonordinal measure.
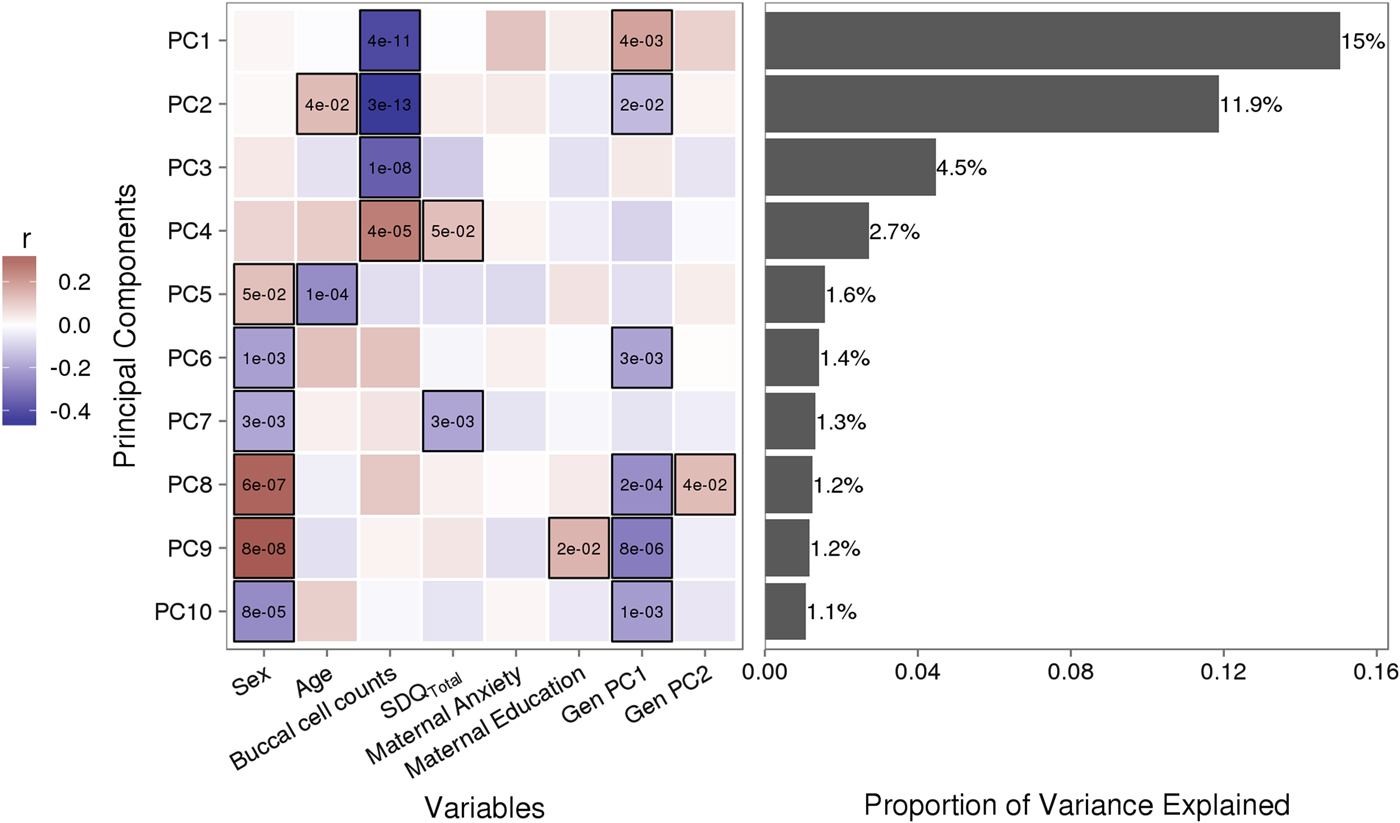
Figure 1. Sources of variation in the DNA methylome. (a) Heatmap describes the bivariate association between variables of interest and the first 10 principal components (PCs) from a principal component analysis of variably methylated probes. The p values are provided for significant associations (p < .05). (b) Bars describe the proportion of variance explained by each principal component. SDQTotal, Strengths and Difficulties Questionnaire total scale score. GenPC1/GenPC2, first and second principal component score from principal component analysis of genetic variation.
The following linear regression model was tested to identify VMPs for which methylation levels were predicted by attachment:
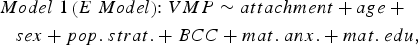 $$\eqalign{Model \; 1 \left(E \; Model \right)\!\! : VMP \sim attachment + age \; + \quad \cr sex + pop. \; strat. + BCC + mat. \; anx. + mat. \; edu\comma \,} $$
$$\eqalign{Model \; 1 \left(E \; Model \right)\!\! : VMP \sim attachment + age \; + \quad \cr sex + pop. \; strat. + BCC + mat. \; anx. + mat. \; edu\comma \,} $$where VMP represents %methylation at a single variably methylated probe; attachment is attachment style, coded into three groups: secure, insecure, and disorganized with secure as the reference category; sex is biological sex of participant, age is age in days at time of buccal sample collection; pop. strat. represents principal components one and two from a PCA of genetic data; BCC is buccal cell composition expressed as a percentage; mat. anx. represents maternal Spielberger State/Trait Anxiety Inventory score at time of biosample collection; and mat. educ. is maternal level of education as a proxy for socioeconomic status.
VMPs that were associated with infant attachment (p < .05) were mapped to genes using Illumina annotation and entered into a Genego Metacore analysis to identify enrichment of biological pathways and processes within our methylation data set. We used MetaCore + MetaDrug version 6.18 (Thomson Reuters, USA).
Genetic contribution to DNA methylation at attachment-associated VMPs
We sought to determine if genetic variation in cis (within 10 kilobases; 10 kb) contributed to variation in DNA methylation at attachment-associated VMPs. We tested for main effects of all SNPs in cis adjusting for relevant covariates (see Model 2). One SNP was tested per model and adjusted for relevant covariates:
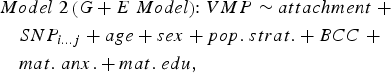 $$\eqalign{& Model \; 2 \left( {G + E \; Model} \right) \!\!: VMP \sim attachment \; + \cr & \quad SNP_{i...j} + age + sex + pop. \; strat. + BCC \; + \cr & \quad mat. \; anx. + mat. \; edu\comma \,} $$
$$\eqalign{& Model \; 2 \left( {G + E \; Model} \right) \!\!: VMP \sim attachment \; + \cr & \quad SNP_{i...j} + age + sex + pop. \; strat. + BCC \; + \cr & \quad mat. \; anx. + mat. \; edu\comma \,} $$where SNPi…j represents the first to the last SNP within a 10-kb window from the attachment-associated VMP.
In parallel, we ran a series of linear models that included an interaction term between infant attachment and each SNP in cis to predict DNA methylation at attachment-associated VMPs (Model 3). One SNP and its corresponding interaction term with infant attachment was tested per model and adjusted for relevant covariates:
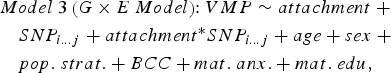 $$\eqalign{& Model \; 3 \left( {G \times E \; Model} \right) \!\!: VMP \sim attachment \; + \cr & \quad SNP_{i...j} + attachment ^{*} SNP_{i...j} + age + sex \; + \cr & \quad pop. \; strat. + BCC + mat. \; anx. + mat. \; edu\comma \,} $$
$$\eqalign{& Model \; 3 \left( {G \times E \; Model} \right) \!\!: VMP \sim attachment \; + \cr & \quad SNP_{i...j} + attachment ^{*} SNP_{i...j} + age + sex \; + \cr & \quad pop. \; strat. + BCC + mat. \; anx. + mat. \; edu\comma \,} $$where attachment* SNP i…j is the interaction term between infant attachment and the first to the last SNP within a 10-kb window from the attachment-associated VMP.
We ranked these additive (G + E) models per VMP to identify the SNP in cis that accounted for the largest proportion of variance in DNA methylation (i.e., highest model adjusted R 2) at a given VMP. Next, we ranked all interaction (G x E) models per VMP to identify the SNP, which in interaction with infant attachment, accounted for the largest proportion of variance in DNA methylation at a given VMP (i.e., highest model adjusted R 2). We selected VMPs for which the top ranked SNP from the additive and interaction model was the same, permitting a direct comparison of models (i.e., a comparison of nested models; Belsky, Pluess, & Widaman, Reference Belsky, Pluess and Widaman2013). We compared adjusted R 2 across each of these three models to identify the model that best explained DNA methylation at attachment-associated VMPs (i.e., the model with the highest adjusted R 2). We also compared our results based on percentage variance explained (adjusted R 2) to a parallel analysis using the Akaike information criterion (AIC), which permits the comparison of nonnested models.
Supplementary analyses
The MAVAN study collected data on a number of measures related to the prenatal period. We used these data to determine if infant attachment buffers the negative effects of prenatal adversity on infant (BSID) and child (SDQ) outcomes, in line with previous reports for different cohort studies (Bergman et al., Reference Bergman, Sarkar, Glover and O'Connor2010; McGoron et al., Reference McGoron, Gleason, Smyke, Drury, Nelson, Gregas and Zeanah2012).
Prenatal adversity
We used a cumulative measure of prenatal adversity described and validated previously within the MAVAN cohort (Silveira et al., Reference Silveira, Pokhvisneva, Parent, Cai, Rema, Broekman and Meaney2017). Information on maternal antenatal illness, birth size, gestational age, household income, lack of material resources, domestic violence, marital strain, maternal anxiety, and maternal depression was combined to form a cumulative score that ranged from 0 to 9. Data on prenatal adversity were available on n = 180 participants included in the subsample of the MAVAN cohort (see Table 1). We used linear regression analysis to test the association between prenatal adversity and (a) infant BSID and (b) child SDQ scores. We also included interaction terms between prenatal adversity and infant attachment classification.
Biological sex-specific effects
We also carried out a series of analyses stratified by biological sex to examine differences in the association between infant attachment and variation in DNA methylation between males and females.
Results
Table 1 describes the characteristics of the children and mothers included in our analysis. There were no significant differences in the age (p = .081) or biological sex (p = .198) of participants across the different attachment classifications.
Infant attachment and child outcomes
One-way ANOVA models showed a significant association between infant attachment and the Mental Development Index, MDI: F (2, 189) = 13.864, p < .001, Cohen's f = .39, and Behavior Rating Scale, BRS: F (2, 192) = 10.74, p < .001, Cohen's f = 0.34, from the BSID at 36 months. Infant attachment was associated with infant psychomotor development (PDI) at trend level, F (2, 190) = 2.385, p = .095, Cohen's f = 0.16. Posthoc analyses of these significant associations revealed differences between the secure and disorganized groups on BRS and MDI scores with large effect sizes (Cohen's d ≥ 0.84), and between the insecure and disorganized groups with medium effect size (Cohen's d ≥ 0.46; BRS: all adjusted p ≤ .026; MDI: all adjusted p ≤ .001). No significant differences were observed between infants with a secure and those with an insecure attachment classification for MDI or BRS scores. These results were largely unchanged following adjustment for child biological sex, population stratification, and maternal education, MDI: F (2, 187) = 12.425, p < .001; BRS: F (2, 190) = 8.679, p < .001; PDI: p > .1.
One-way ANOVA models showed a significant association between infant attachment and child mental health, total SDQ: F (2, 192) = 3.389, p = .036, Cohen's f = 0.19). Post hoc tests revealed significant differences between the secure and disorganized groups (adjusted p = .033) with a medium effect size (Cohen's d = 0.45), with no difference between secure and insecure groups. This association was somewhat reduced following adjustment for child age, biological sex, population stratification, and maternal education, total SDQ: F (2, 190) = 2.654, p = .073. However, when we considered maternal anxiety at the time of SDQ report, to control for potential reporter bias, infant attachment was no longer associated with child total SDQ score (p = .513).
Infant attachment and variation in DNA methylation: Principal components analysis
Figure 1 illustrates the bivariate associations between predictors of interest and the first 10 PC scores from a PCA of VMPs.
Infant attachment was significantly associated with PC2, F (2, 189) = 4.964, p = .008, Cohen's f = 0.24, after adjustment for child age, biological sex, cellular heterogeneity within buccal samples, population stratification, and measures of maternal education and anxiety at time of biosample collection (Figure 2).
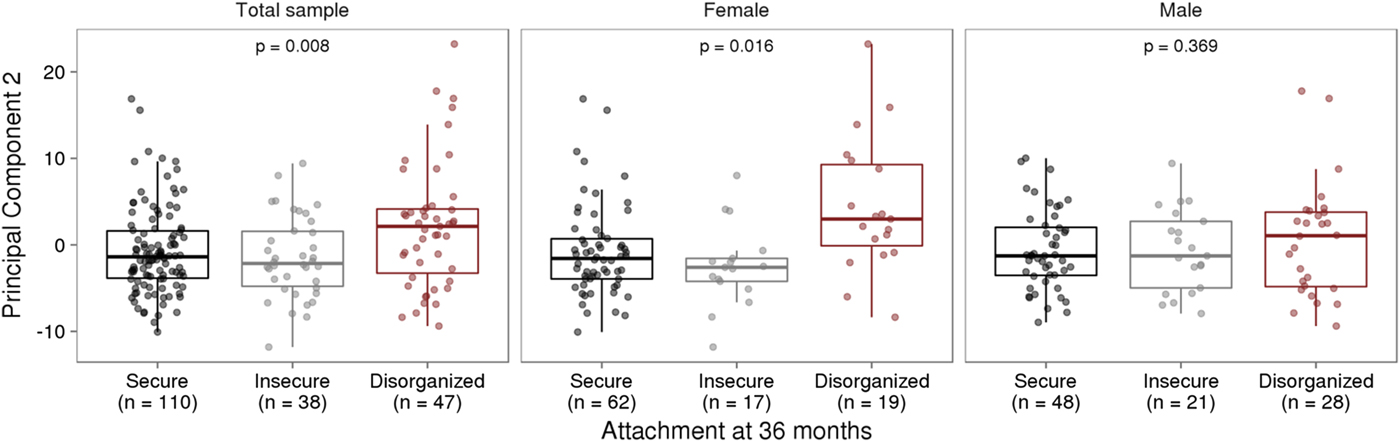
Figure 2. Infant attachment and variation in DNA methylation. (a) Infant attachment associates with principal component two (PC2). PC2 accounts for 11.9% of the variance in DNA methylation of variably methylated probes. (b) Sex-stratified analyses show these effects are evident in females but not in males. The p values are derived from analysis of variance.
Infant attachment and variation in DNA methylation: VMP analysis
No individual VMP passed a Benjamini–Hochberg false discovery rate correction. However, the p value distribution for the model was substantially skewed (Figure 3). A large number of VMPs were associated with infant attachment at a nominal level (unadjusted p = .05). This skewed p value distribution differs from the uniform flat distribution expected under the null hypothesis (i.e., no association between DNA methylation and infant attachment). These differences were most striking for comparisons between secure and disorganized groups (n = 35,761, unadjusted p < .05).
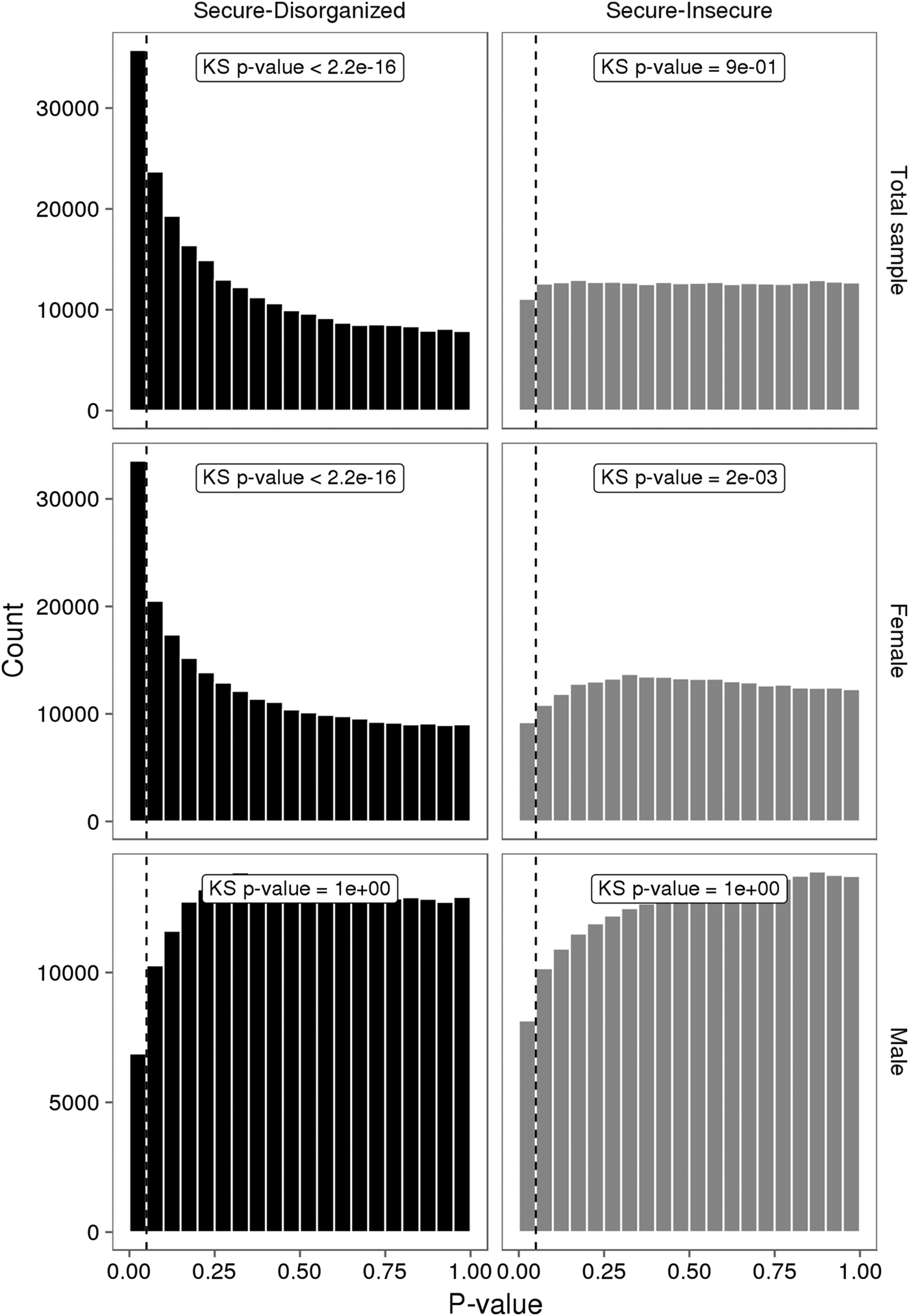
Figure 3. The p value distributions for linear models describing the association between infant attachment and variably methylated probes. The p values derived from one-tailed Kolmogorov–Smirnov (KS) test for uniformity of distribution.
Of note, attachment-associated VMPs (secure vs. disorganized n = 35,761, nominal p < .05) were located in genes for which gene expression has been shown to covary with individual differences in early life experience (Anacker et al., Reference Anacker, Cattaneo, Musaelyan, Zunszain, Horowitz, Molteni and Pariante2013; Klengel et al., Reference Klengel, Mehta, Anacker, Rex-Haffner, Pruessner, Pariante and Binder2013; Pena, Neugut, & Champagne, Reference Pena, Neugut and Champagne2013; Wu et al., Reference Wu, Patchev, Daniel, Almeida and Spengler2014). These included multiple VMPs within the glucocorticoid receptor (NR3C1; cg08845721, cg13648501, cg16586394, cg19457823, cg20753294), estrogen receptor-alpha (ESR1; cg07455133, cg07746998, cg08907436, cg17264271, cg25490334), as well as genes involved in glucocorticoid signaling (SGK1: cg06849960, cg09872934, cg11856561 cg12871835 SGK2: cg21685427, FKBP5: cg07061368, cg07843056 and POMC: cg11894631; see online-only Supplemental Table S.1 for all attachment-associated VMPs).
Pathway analysis of attachment-associated VMPs (nominal p < .05) showed significant enrichment for multiple pathways associated with cell signaling and cell adhesion (Table 2), including the Ephrin signaling pathway.
Table 2. Pathway and process enrichment analysis of attachment-associated variably methylated probes

Notes: Unadjusted p values and p values adjusted for multiple comparisons using the false discovery rate (FDR) are provided.
Child genetic variation contributes to DNA methylation at attachment-associated VMPs
We focused our genetic analysis on attachment-associated VMPs identified from our comparison of secure versus disorganized groups (see Model 1) with at least one SNP in cis (n = 30,965). We identified the SNP from the additive (G + E model; Model 2) or interaction (G x E model; Model 3) models that accounted for the largest proportion of variance in DNA methylation at a specific VMP (see Methods). For 18,042 of these attachment-associated VMPs the same SNP emerged from the additive (Model 2) and interaction model (Model 3). These nested models permitted a direct comparison of adjusted R 2 across models. We observed that the interaction (G x E) model explained the largest proportion of variance (highest adjusted R 2) in DNA methylation for 58.9% of attachment-associated VMPs (see Figure 4). In total, 76.4% of these attachment-associated VMPs benefited from the inclusion of a measure of child genetic variation (Figure 4). We performed a parallel analysis and used AIC to index model fit for prediction models that considered different SNPs across additive and interaction models (i.e., nonnested models) in the prediction of DNA methylation at all attachment-associated VMPs (n = 30,965). These analyses revealed somewhat similar results; model fit for approximately half (48.8%) of all attachment-associated VMPs was improved (i.e., had a lower AIC) when a measure of child genetic variation was considered.
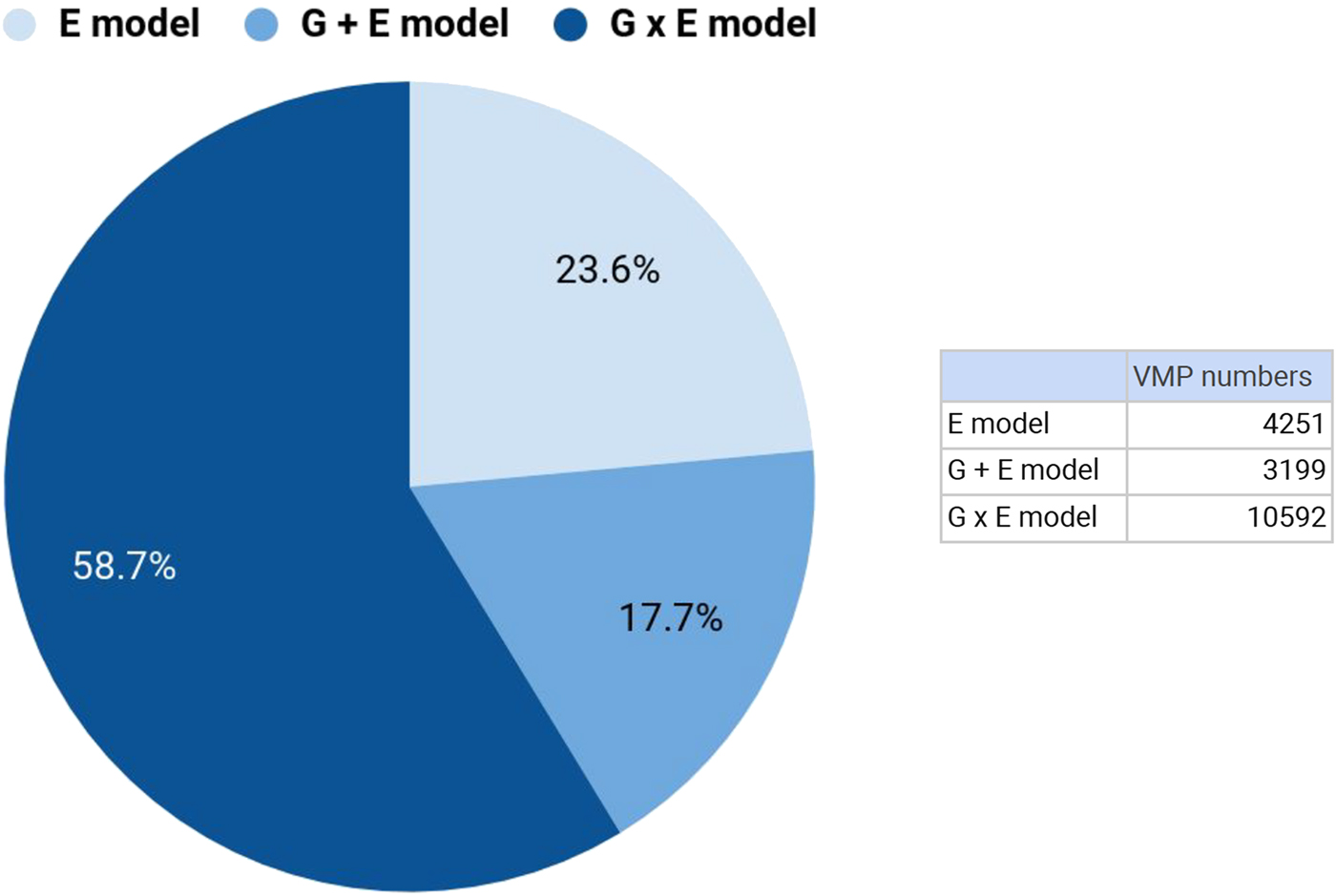
Figure 4. Genetic variation contributes to DNA methylation of attachment-associated variably methylated probes (VMPs). DNA methylation at VMPs were described using linear models that included infant attachment as the environmental predictor (E Model: light blue), the additive effects of infant attachment and a single nucleotide polymorphism (SNP) within 10kb from a VMP (G + E Model: blue), and an interaction model (G × E Model: dark blue) that included infant attachment, a SNP within 10kb from a VMP, and the interaction term between this SNP and infant attachment style. The proportion of variance explained (adjusted R 2) was used to compare models. Percent values on chart denote the number of VMPs that are best explained (highest adjusted R 2) by a specific model, and corresponding numbers of VMPs are provided (see insert). All models were adjusted for buccal cell proportions, population stratification, child age and biological sex, maternal education, and anxiety. kb, kilobase.
Supplementary analyses
Infant attachment and moderation of prenatal adversity effects on child outcomes
Measures of prenatal adversity described previously (Silveira et al., Reference Silveira, Pokhvisneva, Parent, Cai, Rema, Broekman and Meaney2017) and child outcomes were available on a subset of this sample. Linear regression models revealed that prenatal adversity predicted BRS (B = –0.934, p = .018) scores from the BSID at 36 months independent of infant attachment and all covariates. Prenatal adversity was associated with both infant MDI (p = .079) and PDI scores (p = .10) at trend level. We observed a significant interaction between prenatal adversity and attachment security to predict the BRS score, F (2, 153) = 5.82, p = .004, with an effect of prenatal adversity on infant BRS in the disorganized group only (B = –3.14, p < .001). A similar pattern was observed for MDI scores but did not reach statistical significance, F (2, 151) = 2.39, p = .095 (see Figure 5). Prenatal adversity did not interact with infant attachment to predict infant PDI (p > .1).
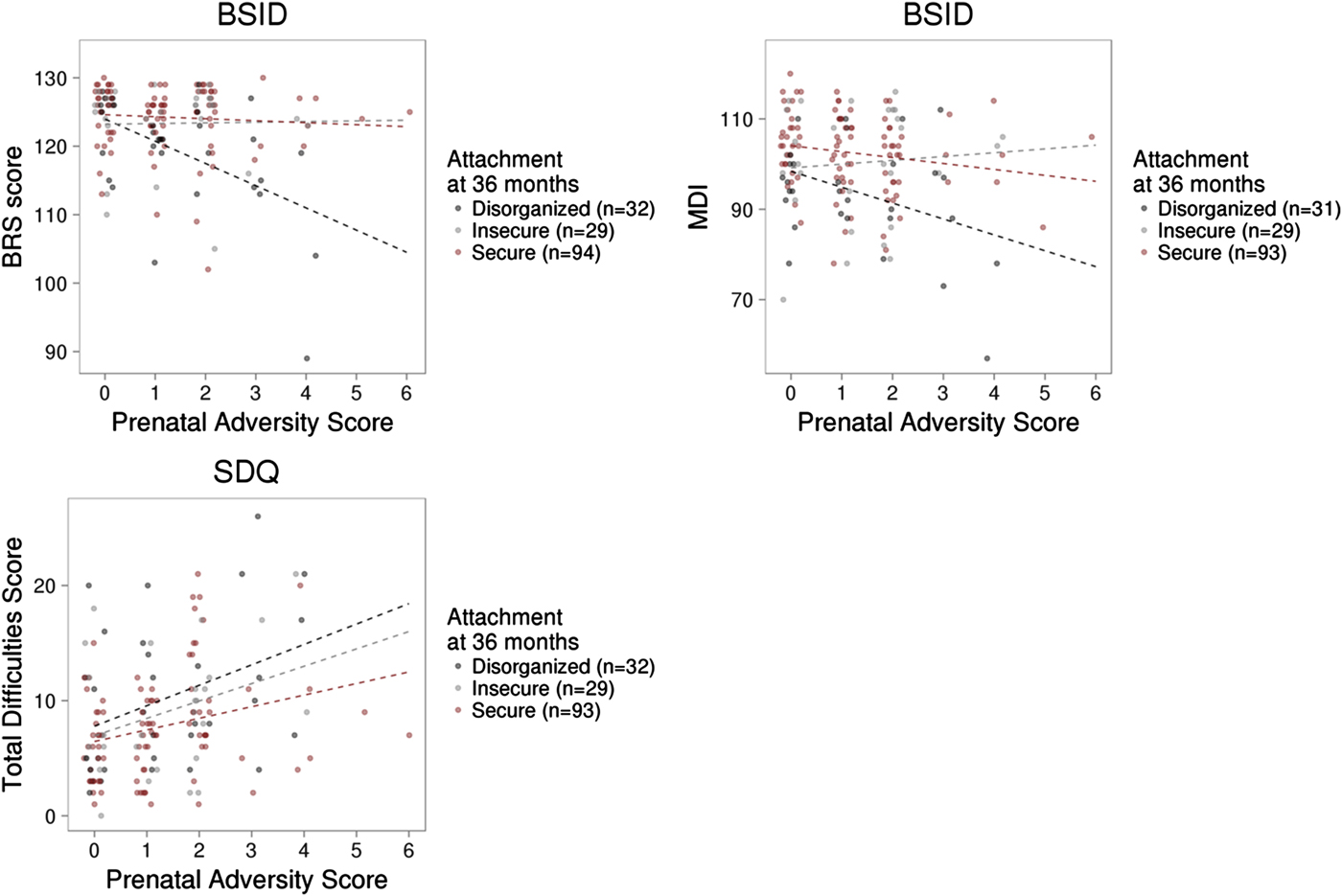
Figure 5. Prenatal adversity, infant attachment, and child outcome. (a) Prenatal adversity interacted with infant attachment to predict the Behavior Rating Scale score of the Bayley Scale of Infant Development (BSID) at 36 months. (b) The interaction between prenatal adversity and infant attachment did not significantly predict the Mental Development Index of the BSID, or (c) total difficulties from the Strengths and Difficulties Questionnaires in middle childhood (mean age = 6.99, SD = 1.33 years).
Prenatal adversity predicted child total SDQ score (B = 0.71, p = .028) independent of all covariates considered in our SDQ model, including measures of infant attachment and maternal trait anxiety (see Methods). However, prenatal adversity did not interact with infant attachment to predict child total SDQ scores. (See Supplemental Table S.2 for all prenatal adversity linear regression results).
Sex-specific effects of infant attachment on DNA methylation of attachment-associated VMPs
We carried out exploratory analyses to examine potential sex differences in the association between infant attachment and variation in DNA methylation. Analyses stratified by biological sex of our PCA of VMPs revealed a significant association between PC2 and infant attachment in females but not in males (Figure 2). Similar results were observed from our analyses of infant attachment and DNA methylation at individual VMPs. Differences in DNA methylation between secure and disorganized classifications were most pronounced in females but not in males (see Figure 3).
Discussion
We describe the association between infant attachment at 36 months of age and DNA methylome variation in childhood. To our knowledge this is the first and largest assessment of infant attachment and variation in DNA methylation carried out to date. These findings contribute to a growing literature describing the biological embedding of early experience and suggest a persisting influence of infant attachment on variation in DNA methylation. Our study also provides the first step toward characterizing a molecular signature of infant attachment, which may inform early interventions that seek to promote attachment security.
Our PCA of VMPs showed that infant attachment associates with a significant portion of the variance across the DNA methylome. These data extend those of Moore et al. (Reference Moore, McEwen, Quirt, Morin, Mah, Barr and Kobor2017), and suggest normal variations in the quality of the early caregiving environment may influence variation across the DNA methylome.
Across both our pathway and process enrichment analyses, we observed enrichment for genes related to cell adhesion/cell migration, such as the Ephrin signaling pathway. Ephrins, which bind tyrosine kinase Eph receptors, are critically involved in neuronal interaction, cell migration, and cell differentiation. This is the first report in humans documenting an association of the early care environment and DNA methylation of Ephrin family members; however, our findings draw support from a number of preclinical models. For example, Provençal et al. (Reference Provençal, Suderman, Guillemin, Massart, Ruggiero, Wang and Szyf2012) report differential DNA methylation of Ephrin family members (ENFB2/ENFA5) in T cells and prefrontal cortex samples from nonhuman primates exposed to their peer-rearing model of early adversity (Provençal et al., Reference Provençal, Suderman, Guillemin, Massart, Ruggiero, Wang and Szyf2012). Similarly, female Ephrin knock-out mice (Ephrin-A5−/−) exhibit reduced levels of maternal care toward their offspring (Sheleg et al., Reference Sheleg, Yu, Go, Wagner, Kusnecov and Zhou2017). Together with our own data, these findings serve to highlight Ephrin signaling as a novel pathway both sensitive to the effects of the early care environment, and potentially, for promoting caregiving behavior.
We also observed significant enrichment for POMC processing within attachment-associated VMPs. However, we note that this enrichment derives in part from the large number of protein products, which arise from the cleavage of POMC (Pritchard & White, Reference Pritchard and White2007; Wu et al., Reference Wu, Patchev, Daniel, Almeida and Spengler2014). The methylation of the Pomc promoter is influenced by variation in maternal care (Wu et al., Reference Wu, Patchev, Daniel, Almeida and Spengler2014), and POMC is centrally involved in glucocorticoid signaling. We also observed differential methylation for a number of VMPs within the glucocorticoid receptor NR3C1 and glucocorticoid-sensitive targets, including FKBP5 and SGK1, across attachment groups. SGK1 gene expression is increased in blood samples from depressed individuals and also within the hippocampus of prenatally stressed rats (Anacker et al., Reference Anacker, Cattaneo, Musaelyan, Zunszain, Horowitz, Molteni and Pariante2013). Klengel et al. (Reference Klengel, Mehta, Anacker, Rex-Haffner, Pruessner, Pariante and Binder2013) report altered FKBP5 methylation and glucocorticoid sensitivity following exposure to childhood trauma that covaries with hippocampal volume as well as with clinical symptoms and treatment outcomes in posttraumatic stress disorder (Yehuda et al., Reference Yehuda, Daskalakis, Desarnaud, Makotkine, Lehrner, Koch and Bierer2013).
We also found that infant attachment was associated with variation in DNA methylation at multiple sites within ESR1, which encodes estrogen receptor alpha (ERα). Unfortunately, we were unable to quantify the effects of infant attachment and associated differential methylation of ESR1 on its gene expression. Nevertheless, the variation in ESR1 methylation as a function of attachment is consistent with studies showing that the frequency of pup licking/grooming in the rat predicts DNA methylation of an Esr1 promoter and Esr1 expression in the hypothalamus and amygdala (Champagne & Meaney, Reference Champagne and Meaney2006; Pena et al., Reference Pena, Neugut and Champagne2013). Binding of ERα to its consensus binding site can influence DNA methylation of target genes (Guintivano, Arad, Gould, Payne, & Kaminsky, Reference Guintivano, Arad, Gould, Payne and Kaminsky2013; Kangaspeska et al., Reference Kangaspeska, Stride, Metivier, Polycarpou-Schwarz, Ibberson, Carmouche and Reid2008; Metivier et al., Reference Metivier, Gallais, Tiffoche, Le Peron, Jurkowska, Carmouche and Salbert2008) consistent with transcription factor-mediated remodeling of the DNA methylome (Stadler et al., Reference Stadler, Murr, Burger, Ivanek, Lienert, Scholer and Schubeler2011; Weaver et al., Reference Weaver, D'Alessio, Brown, Hellstrom, Dymov, Sharma and Meaney2007).
The association between infant attachment and methylation of ESR1 is of interest considering the known association between the early life experience and the timing of female sexual maturation. The quality of parent–child relations in humans (Ellis & Essex, Reference Ellis and Essex2007; Ellis, Shirtcliff, Boyce, Deardorff, & Essex, Reference Ellis, Shirtcliff, Boyce, Deardorff and Essex2011), including attachment insecurity (Belsky & de Haan, Reference Belsky and de Haan2011), associate with the timing of female sexual maturation, consistent with life-history theories of early programming of human reproductive strategy under conditions of adversity (Chisholm, Reference Chisholm1996). While the precise mechanism linking early family life to the timing of sexual maturation is unknown, there is evidence from Gene × Environment studies showing that the influence of early family life on the timing of menarche is moderated by ESR1 variants (Hartman, Widaman, & Belsky, Reference Hartman, Widaman and Belsky2015; Manuck, Craig, Flory, Halder, & Ferrell, Reference Manuck, Craig, Flory, Halder and Ferrell2011). It will be of interest to determine if attachment-associated differences in ESR1 DNA methylation covary with pubertal timing in females. Likewise, it will be important to determine if an altered rate of female sexual maturation contributes to the marked sex differences we observe in the association between infant attachment and variation in DNA methylation across groups.
Our findings highlight the genome as an important source of variation in DNA methylation of attachment-associated VMPs, consistent with a large literature on genetic effects on DNA methylation (Hannon et al., Reference Hannon, Spiers, Viana, Pidsley, Burrage, Murphy and Mill2016; Jaffe et al., Reference Jaffe, Gao, Deep-Soboslay, Tao, Hyde, Weinberger and Kleinman2016; Ng et al., Reference Ng, White, Klein, Sieberts, McCabe, Patrick and De Jager2017). Models that considered child genetic variation provided the best fit (adjusted R 2 or AIC) for approximately half of the attachment-associated VMPs. These findings are consistent with those of Teh et al. (Reference Teh, Pan, Chen, Ong, Dogra, Wong and Holbrook2014), who report that the majority of variably methylated regions identified in umbilical cord samples from Singaporean neonates were best explained by gene–environment interactions. Such findings further highlight the need to consider genetic variation when seeking to characterize the biological embedding of early life experience.
In line with previous studies, we found that infant attachment predicted infant cognitive development (Bergman et al., Reference Bergman, Sarkar, Glover and O'Connor2010) but also predicted child behavior during this mildly stressful novel stimulus (i.e., the BSID). The association between infant attachment and both MDI and BRS scores represented medium to large effects. Consistent with Bergman et al., we also observed an interaction between a measure of prenatal adversity and infant attachment style to predict BSID scores. However, we note that unlike Bergman et al., our results were restricted to the BRS. It is possible that differences in the age of assessment (17 months vs. 36 months) and our use of a three-group attachment classification system contributed to differences across our studies. However, we do note that the interaction between prenatal adversity and infant attachment is directionally consistent with the previous report (Bergman et al., Reference Bergman, Sarkar, Glover and O'Connor2010). As such, our data provide further evidence that attachment security may buffer the effects of prenatal adversity on measures of child outcome (Bergman et al., Reference Bergman, Sarkar, Glover and O'Connor2010), which may be of relevance for later psychopathology (McGoron et al., Reference McGoron, Gleason, Smyke, Drury, Nelson, Gregas and Zeanah2012).
We did not observe a significant association between infant attachment and child total mental health symptoms. Our data highlight the importance of adjusting for maternal mood when considering measures of child outcome based on maternal report. Likewise, we did not observe an interaction between prenatal adversity and infant attachment to predict child SDQ. It is possible that our choice of measure (SDQ) contributed to this null finding. In their meta-analysis Fearon et al. (Reference Fearon, Bakermans-Kranenburg, van IJzendoorn, Lapsley and Roisman2010) found a stronger association between infant attachment and child externalizing symptoms when assessed through observational methods rather than maternal report.
There are important limitations to this study. First, although this is the largest pediatric study of infant attachment and DNA methylation carried out to date, none of the individual VMPs associated with attachment were significant following adjustment for genome-wide correction. This is not surprising in light of the relatively small effect sizes observed between psychosocial adversity and variation in DNA methylation. Few studies in the field of biological psychiatry report changes in DNA methylation greater than 10%, despite functional effects of such changes on gene expression (Mehta et al., Reference Mehta, Klengel, Conneely, Smith, Altmann, Pace and Binder2013). Our analysis was intended to determine whether infant attachment at 36 months of age was a significant source of interindividual variation in genome-wide DNA methylation. We also attempted to limit false-positive findings by including only those probes that showed the interindividual differences (i.e., VMPs). Likewise, due to our use of a three-group classification for infant attachment, our secondary analyses, which considered interactions with prenatal adversity and biological sex, are likely underpowered. As such, these data should be considered preliminary until replicated in an independent sample.
Second, the correspondence between buccal and neuronal methylation profiles is largely unknown. However, it is possible that regions of the genome that are sensitive to steroid hormones, which readily pass the blood–brain barrier, may produce coordinated modifications of DNA methylation across multiple tissues. The increasing availability of publicly available data sets and online tools (Edgar, Jones, Meaney, Turecki, & Kobor, Reference Edgar, Jones, Meaney, Turecki and Kobor2017), which examine the concordance of DNA methylation across peripheral and central tissues, will facilitate the interpretation of findings from studies that use surrogate tissues for brain-based phenotypes.
In summary, our data show that the early care environment covaries with variation in genome-wide DNA methylation in middle childhood. The study of the DNA methylome provides a direct measure of the potential impact of the social environment at the level of the individual child and suggests a persisting influence of infant attachment on the DNA methylome. A deeper understanding of the molecular architecture of infant attachment will help monitor the efficacy of attachment-based therapeutic interventions, which seek to mitigate the lasting effects of early adversity on child development.
Supplementary Material
To view the supplementary material for this article, please visit https://doi.org/10.1017/S0954579418000627.