


DISCUSSION QUESTIONS
-
• Can Mg battery compete with current or future Li-ion technology?
-
• The urgent requirement in Mg battery cathode research.
-
• How to utilize the advantage of metal Mg anode while avoiding challenges on the cathode side?
Introduction
The automobile industry is facing great challenges associated with the rising costs of oil and gas, as well as the growing crisis about the environmental pollution caused by traditional engines. It is therefore motivated to put much research resources on novel mobility concepts. The first vehicle that utilized a hybrid energy source of gas and electricity was marketed by Toyota Prius in 1997. Entering the 21st century, new vehicle concepts such as plug-in hybrid or purely electrical vehicle have emerged as potential future mobility, all of which strongly rely on the development of novel sources for energy storage and transportation.
Coincidental with the appearance of these novel mobility concepts, we have evidenced the mature of Li-ion battery technology in the past decades, which powers the rapid boost of portable electronic devices. Li-ion battery has also been used in several marketed electronic vehicles. Yet in large scale applications where the size of the battery pack increases, the continuous improvement of the energy density and power density of Li-ion battery remains challenging. Besides, the limited storage of lithium minerals in a few countries indicates a potential risk to the supply chain of lithium in a large scale. Reference Tarascon and Armand1 These issues have motivated the research of electrochemical systems that utilize the shuttle of more earth-abundant ions, including monovalent Na+, Reference Palomares, Serras, Villaluenga, Hueso, Carretero-Gonzalez and Rojo2 bivalent Mg2+, Reference Yoo, Shterenberg, Gofer, Gershinsky, Pour and Aurbach3 and even trivalent Al3+. Reference Lin, Gong, Lu, Wu, Wang, Guan, Angell, Chen, Yang, Hwang and Dai4
The current Review deals with one of such post Li-ion battery candidates, rechargeable magnesium battery. Compared with that of lithium, magnesium electrochemistry certainly has several advantages. Table 1 lists several key properties of lithium and magnesium. The crust abundance of magnesium is about three orders of magnitude higher than that of Li. More importantly, magnesium minerals widely exist in the lithosphere, suggesting the least concern about its supply risk. The market price of magnesium is $2700/ton, about 4% of lithium, $64,800/ton, while the annual production of magnesium in 2012 is about 20 times higher than that of lithium. These economic merits suggest that magnesium battery has better potential to become a more cost friendly technology than current Li-ion battery. Considering the wide availability of magnesium minerals, it may even be postulated that magnesium battery could potentially become the choice for grid energy storage in future, where the cost and availability of materials are of great concern.
Table 1. Key economical and electrochemical properties of lithium and magnesium.
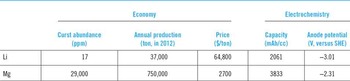
The electrochemical property of Mg is also promising. Because of the bivalency of Mg2+ ions, the specific volumetric capacity of metal Mg anode is 3833 mAh/cc, which is nearly twice of that for Li metal. Reference Matsui5 The deposition and dissolution on metal Mg anode is also able to exhibit high Columbic efficiencies in appropriate electrolytes, which in some cases was close to 100%. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6–Reference Liu, Shao, Li, Gu, Hu, Xu, Nie, Chen, Wang and Liu8 More importantly, it is well-known that the electrochemical deposition of Li typically forms dendritic morphology, which prevents the direct usage of metal Li anode. Current Li-ion battery uses graphite as the anode, which further decreases the capacity to 740 mAh/cc (assume density 2 g/cc). The electrochemical deposition of magnesium, on the other hand, showed different characteristics. It preferably developed flat surfaces instead of the dendrites. Reference Matsui5,Reference Ling, Barnejee and Matsui9,Reference Jäckle and Groß10 Therefore, metal Mg can serve as a safe choice for anode.
The first prototype rechargeable Mg battery was reported by Aurbach's group in 2000. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6 They brilliantly married metal Mg anode and Chevrel phase (CP) Mo6S8 cathode in a Grignard based electrolyte. Since then, the research of Mg battery has been carried out worldwide with increasing interest. However, compared to current Li-ion battery, Mg battery is still in its infant stage. Among many challenges that must be solved before any possible commercialization, searching cathode candidates with high energy and power density lies in the center of Mg battery development. Reference Yoo, Shterenberg, Gofer, Gershinsky, Pour and Aurbach3 The CP cathode only delivers a voltage of ∼1.2 V and a capacity of ∼120 mAh/g. Thus at that time it was only proposed as an alternative to Ni–Cd battery technology. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6 Although cathodes with higher voltages or capacities were reported recently, we have to admit the fact that there is still no cathode candidate that meets the request for commercial-ready usage. Reference Yoo, Shterenberg, Gofer, Gershinsky, Pour and Aurbach3
Toyota has entered the research of Mg battery since 2008. After extensively working on this area for more than 6 years, in coincidence with the rapid increase of the interest in this field, we feel now it is appropriate timing to summarize the reported literature and give our perspective for future development of Mg battery. In the current Review, the reported performance of Mg battery cathode is summarized and compared. It should be noted that the efforts on the cathode research were mostly carried out in conventional electrolytes (Mg(TFSI)2 or Mg(ClO4)2 in acetonitrile or propylene carbonate) before the seminal report of Aurbach et al. in 2000. However, due to the lack of the knowledge of Mg deposition/extraction in convention electrolytes, some controversies seemed to exist. Now most cathode research rely on the usage of Grignard based electrolytes. To avoid confusion, the current Review only includes the reports after 2000. For readers who are interested in the work before 2000, we refer them to the review of Novak et al., which excellently summarized the work done in that period. Reference Novak, Imhof and Haas11
To provide a broad and comprehensive review as much as possible, we define the scope of current Review to batteries that utilize the dissolution and deposition of Mg on the anode during the discharge and charge process. With this definition, the cathode reaction no longer necessarily involves the participation of Mg2+ ions in certain cell configurations. As we will discuss later, this broader Mg battery concept enables the fabrication of cells with better rate performance when the cathode reaction does not involve sluggish Mg migration.
It is important to note that the electrochemical measurement of the performance of Mg battery cathode is strongly affected by the choice of other battery components in the test. For instance, if the cell is operated in conventional electrolytes with metal Mg anode, the passivation on the anode surface kills any recorded electrochemical activity. Reference Singh, Arthur, Ling, Matsui and Mizuno12 In such circumstance, the cell performance is limited by the deposition and dissolution of metal Mg on the anode instead of any cathode reaction. Reference Mizuno, Singh, Arthur, Fanson, Ramanathan, Benmayza, Prakash, Liu, Glans and Guo13 Besides, Grignard electrolyte containing halide species can be corrosive to other parts of the cell such as current collectors. Reference Muldoon, Bucur, Oliver, Sugimoto, Matsui, Kim, Allred, Zajicek and Kotani14,Reference Muldoon, Bucur, Oliver, Zajicek, Allred and Boggess15 Therefore, the usage of coin cell with parts that are not resistive to the corrosion will generate biased electrochemical data if these side reactions are not seriously taken into account. Despite their importance, a comprehensive discussion about the anode, electrolyte, and the current collector would be out of the scope of the current Review. We recommend the readers who have great interest in these fields to Ref. 16. Reference Muldoon, Bucur and Gregory16
The whole Review is organized as the following. In the section “Performance review of Mg battery cathodes,” the reported literature about Mg battery cathode is reviewed, categorized by their chemical composition. It includes the CP, oxides, polyanions, chalcogenides, oxygen, sulfur, and other types of cathodes. In the section “Summary and perspective,” we provide a summary about the current status of Mg battery cathode research and highlight challenges that must be solved. A perspective is provided after that with suggestion for possible research directions in this field. Conclusive remarks are given in the section “Conclusion.”
Performance review of Mg battery cathodes
Chevral phase
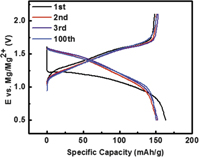
The cathodes currently used in Li-ion rechargeable battery are mostly operated based on the intercalation/de-intercalation chemistry. The same concept has also been attempted to explore cathode materials for Mg batteries. However, despite numerous materials having been investigated, only one group of materials named as CP showed excellent cyclability and relative high rate capability. The general formula of CP is M x Mo6T8, where M = metal, e.g., Li, Mg, Cu, Zn, etc., and T = S, Se, and Te. In their seminal paper, Aurbach et al. reported CP-Mo6S8 as reversible cathode material in Mg battery with the cell configuration of Mg/Grignard electrolyte/CP. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6 Figure 1(a) shows a typical coin cell cycling curve using CP Mo6S8 as cathode material. The initial discharge capacity reached ∼80 mAh/g at a 0.3 mA/cm2 using the electrolyte of 0.25 M Mg(AlCl2BuEt)2 in tetrahydrofuran (THF). Two voltage plateaus were observed at 1.3 and 1.1 V during discharge process. The most remarkable performance of this cell was its excellent cyclability. In their report, the cathode sustained more than 2000 charge–discharge cycles at 100% depth of discharge at practical rates (0.1–1 mA/cm2) in a wide temperature range (−20 to 80 °C), with capacity fading of less than 15%. Typical cycling performance was shown in Fig. 1(b).

Figure 1. (a) Electrochemical behavior and the basic structure of the CP cathode. (b) Cycling performance of CP at a constant current of 0.3 mA/cm2. The inset shows the voltage profile at certain cycle. Reprinted with permission from Ref. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6. Copyright 2000 Nature Publishing Group.
The mechanism of the intercalation/de-intercalation of Mg2+ ions in CPs has been intensively studied. Reference Levi, Lancry, Mitelman, Aurbach, Ceder, Morgan and Isnard17–Reference Levi, Levi, Chasid and Aurbach21 The crystal structure of CPs consists of octahedral clusters of Mo (Mo6) inside cubic anion frameworks as stacks of Mo6T8 blocks within a three-dimensional open structure as shown in the insertion of Fig. 1(a). Mo6 cluster exhibits variable valence during the insertion and removal of Mg2+ ions, while the anionic framework is flexible with multidirectional paths for ionic diffusion. CP-Mo6T8 can theoretically sustain up to four electrons exchanging, which corresponds to the insertion of two bivalent Mg2+ ions. This unusual structure provides a large number of close vacant sites and the metallic Mo6 clusters. Especially the Mo6 clusters is speculated as a critical factor to ensure high Mg2+ diffusivity in solid state host because they can easily compensate the charge-unbalance due to the introduction of bivalent Mg2+ ions. Reference Levi, Gofer and Aurbach20
The synthesis of CPs typically starts with the preparation of Cu2Mo6S8 followed by leaching out Cu2+ ions. Cu2Mo6S8, chemically leached in I2/acetonitrile or in HCl/H2O, was able to accommodate the theoretical amount of Mg2+ ions upon the first discharge of the electrode (2 Mg atoms per formula unit). Reference Lancry, Levi, Gofer, Levi, Salitra and Aurbach22 A specific capacity of 90–100 mAh/g was achieved with excellent stability upon long-term cycling. Kim et al. reported an effective method to control the composition of CP-Cu x Mo6S8 by adopting a chemical intercalation process. Reference Woo, Yoo, Cho, Park, Kim, Kim, Kim and Kim23 A high reversible capacity close to the theoretical value was achieved at the Cu1.3Mo6S8 electrode with good discharge rate capability (1 C with 90% capacity retention) and cycling performance (0.05 C over 50 cycles) at room temperature. Recently, this group further investigated the reaction mechanism of Cu doped CP. Reference Choi, Kim, Woo, Cho, Choi, Choi, Lee, Park and Kim24 They found that Cu metal-like shell formed during Mg2+ ions intercalation and this Cu shell disappeared as Mg deintercalation. This finding helped elucidate the origin of the additional capacity of the Mo6S8 cathode arising from Cu addition and improve the electrochemical performance of the Mo6S8 cathode for rechargeable Mg batteries.
Substituting Se for S in CP improved the capacity of the first discharge plateau, while negligibly affecting the second discharge plateau. Reference Levi, Lancry, Mitelman, Aurbach, Isnard and Djurado18 The initial capacity of Mo6S6Se2 reached 110 mAh/g. Impressively, the rate behavior was improved for Se substituted CP. At 1 C rate, the capacity of Mo6S6Se2 decreased only by 10%, compared to the optimal value at 0.1 C rate. For comparison, the 1 C capacity of nonsubstituted Mo6S8 dropped to only half of the capacity at 0.1 C. The presence of Se in the anionic framework increased its polarizability and thus improved solid state diffusion of Mg2+ ions. Besides, the presence of Se changed the local geometry of Mg insertion sites and enhances Mg migration.
Typically, CPs are synthesized by high temperature solid state reactions of elemental blend of copper, molybdenum, and sulfur powders at ∼1423 K for 7 days or by a molten salt route using Mo–MoS2–CuS reactants in a KCl salt, and heat treating the reaction mixtures at ∼1123 K for 60 h in Ar atmosphere. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6 The as-prepared materials were leached with acid to remove Cu. Both methods are very time consuming and most importantly the as-synthesized particle is still within micro size range, which limits the rate performance. Ryu et al. investigated the relationship between Mo6S8 particle size and the ratio of KCl salt with precursors. Reference Ryu, Park, Cho, Kim and Kim25 Increasing the ratio of KCl melting salt from 1:1 to 4:1 decreased the particle size of CP from 0.75 to 0.24 μm. Consequently, the initial discharge capacity changed from 73 to 85 mAh/g. Saha et al. also tried to control the particle size through a co-precipitation method followed by a conventional calcination process. Reference Saha, Jampani, Datta, Okoli, Manivannan and Kumta26 The particle size of the target material decreased to ∼500 nm and a capacity over 60 mAh/g was observed at a rate up to 1.5 C. Cheng et al. used a facile process to precipitate the precursors on graphene to increase the conductivity of the final material and control its particle size. Reference Cheng, Parent, Shao, Wang, Sprenkle, Li and Liu27 Using this method, Mo6S8 showed better rate performance (2 C at a capacity of 70 mAh/g) and cycling behavior (retain the capacity over 70 mAh/g over 150 cycles).
Despite the fascinating cyclability and high Mg mobility, the voltage of CPs is only ∼1.0 to ∼1.2 V against Mg/Mg2+. The utilization of a heavy transition metal cluster (Mo6) also limits its capacity to less than 150 mAh/g. These facts suggest that CPs can only be considered as moderate or low energy density cathodes. In their original paper, Mg battery based on CP cathode was only suggested to be an alternative to Ni–Cd battery. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6 Other cathode candidates with higher voltage and/or capacity must be pursued if Mg battery is competing with Li-ion technology. However, Mo6S8 is still considered as the best-known model cathode for magnesium battery and will continue to serve as the baseline system to evaluate the suitability of new magnesium electrodes and electrolytes.
Other cluster-type cathodes
A crucial factor that leads to the nice performance of CPs is the unique Mo6 cluster contained in the structure. Levi et al. argued that the existence of a cluster-like redox center enables the fast electron re-distribution during the migration of guest Mg2+ ions. Reference Levi, Gofer and Aurbach20 The local electron neutralization requests the distribution of two electrons simultaneously with the migration of Mg2+, which in classical intercalation-type cathode is kinetically sluggish. For CPs, however, the electron re-distribution only changes the formal charge of individual Mo ions by 1/3e. This greatly lowers the barrier for the electron re-distribution and consequently improves Mg2+ migration. Accordingly, they proposed that other materials that contain redox active clusters may also serve as Mg cathode candidates.
We recently reported another cluster-type cathode material that entirely consists of nonmetal ions, the fullerene (C60). Reference Zhang, Mizuno and Ling28 The C60 cathode material contains a group of carbon atoms bonding together by interatomic forces to form clusters. The electrochemical performance of C60 cathode was shown in Fig. 2(a). Reference Zhang, Mizuno and Ling28 Two voltage plateaus at 1.4 and 1.1 V were observed. The initial capacity reached ∼50 mAh/g at the cut off voltage of 0.8 V. The most impressive performance of C60 cathode was the remarkable rate capability compared to other Mg battery cathodes. Figure 2(b) shows the rate performance of C60 cathode. Even at the current density of ∼1.5 mA/cm2, the C60 cathode still retained 44% of its capacity operated at 0.019 µA/cm2. The excellent electrochemical performance of C60 was related to its cluster structure which effectively delocalizes extra electrons to the whole cluster rather than on one or two atoms.
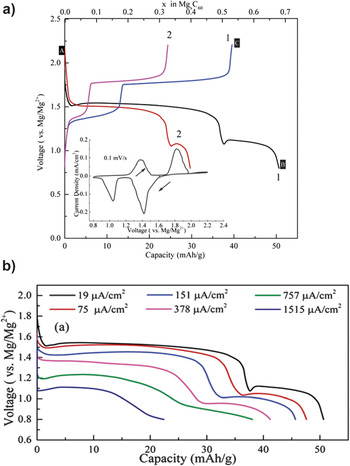
Figure 2. (a) Voltage profiles of C60 cathode at the current density of 19 μA/cm2. The inset shows the cyclic voltammetry curve. (b). Discharge curves of C60 cathode at different current densities. Reprinted with permission from Ref. Reference Zhang, Mizuno and Ling28. Copyright 2015 Royal Society of Chemistry.
Taniguchi et al. reported another cluster based material, Mo9Se11 as cathode material for Mg battery. Reference Taniguchi, Yoshino, Gu, Katsura and Takagi29 Similar to CP, its structure contains Mo9 clusters. The electrochemical test was carried out using Mo9Se11 cathodes and magnesium metal anodes in 0.25 M Mg(AlCl2EtBu)2/THF electrolyte solution. The derivative of the capacity with respect to the potential showed two distinct peaks for the cathodic process at 1.0 and 0.94 V, and the anodic process at 1.18 and 1.1 V, respectively. Because of the high molecular weight of Mo9Se11, the reversible capacity was less than 50 mAh/g.
These results suggest that cluster based materials have great potential to serve as Mg battery cathodes. Further investigation is necessary to better explore this area both theoretically and experimentally.
Oxide cathodes
Oxides, including MnO2, V2O5, MoO3, and spinels, have been intensively studied as Mg battery cathodes. Manganese dioxide (MnO2) has been investigated in Li-ion batteries due to its environmentally benign properties and low cost synthesis process date back to 1980s. MnO2 has many polymorphic forms, including several open channel structures (α phase: 2 × 2 channel; β-phase: 1 × 1 channel; γ-phase: 1 × 2 channel), layered δ phase and spinel phase. These phases have been found to be suitable for the insertion of various cations with different sizes. In the last 3 years, α-MnO2 has been intensively studied in our group as a cathode material for rechargeable Mg battery. This material showed a high capacity up to 280 mAh/g (Fig. 3) at the first discharge cycle using hexamethyldisilazide magnesium chloride (HMDS–MgCl) as electrolyte. Reference Zhang, Yu, Nam, Ling, Arthur, Song, Knapp, Erlic, Yang and Matsui30 The Columbic efficiency of the first cycle exceeded 100%, possibly due to side reactions in the charge process such as Mg deposition and electrolyte decomposition. The reversible magnesiation/demagnesiation was proven by x-ray photoelectron spectroscopy (XPS) and x-ray absorption spectroscopy (XAS) of Mn states. However, the pristine state was not recovered after the battery was recharged to 3.0 V, suggesting the electrochemical reaction was not completely reversible. In addition, the discharge capacity quickly dropped to only half of the initial value and further faded at the following cycles. Similar observation was reported by Rasul et al. They prepared the K+ ion stabilized α-MnO2 through hydrothermal method. When testing the performance of K+ ion stabilized α-MnO2 in a three-electrode cell using 1 M Mg(ClO4)2–acetonitrile solution as electrolyte, the discharge capacity quickly dropped in the second cycle. The capacity of α-MnO2 was increased by adding carbon black during hydrothermal synthesis. However, the capacity fading issue was not significantly improved.
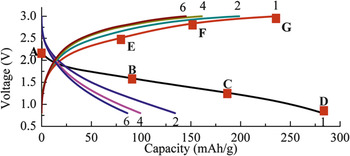
Figure 3. Galvanostatic cycle of α-MnO2 at 19 μA/cm2. Reprinted with permission from Ref. Reference Zhang, Yu, Nam, Ling, Arthur, Song, Knapp, Erlic, Yang and Matsui30. Copyright 2012 Elsevier.
To understand the reaction mechanism between α-MnO2 and Mg, advanced analysis about the discharge product was carried out using high resolution transmission electron microscopy (HR-TEM) and soft x-ray absorption spectroscopy (sXAS). Reference Arthur, Zhang, Ling, Glans, Fan, Guo and Mizuno31 During the magnesiation (discharge), an amorphous layer was formed on the surface of α-MnO2 while the core still remained its crystallinity. Electron energy loss spectroscopy and sXAS analysis revealed that the amorphous shell consisted of reduced Mn3+, Mn2+ as well as Mg2+ species. Therefore, we interpret the amorphous product as gradually reduced manganese oxides mixed with magnesium oxide. The proposed reaction mechanism follows Reference Arthur, Zhang, Ling, Glans, Fan, Guo and Mizuno31
 $${\rm{M}}{{\rm{g}}^{2 + }} + 2{\rm{Mn}}{{\rm{O}}_2} + {e^ - } \to \left( {{\rm{Mg}},{\rm{Mn}}} \right){\rm{O}}\left( {{\rm{shell}}} \right) + {\rm{Mn}}{{\rm{O}}_2}\left( {{\rm{core}}} \right).$$
$${\rm{M}}{{\rm{g}}^{2 + }} + 2{\rm{Mn}}{{\rm{O}}_2} + {e^ - } \to \left( {{\rm{Mg}},{\rm{Mn}}} \right){\rm{O}}\left( {{\rm{shell}}} \right) + {\rm{Mn}}{{\rm{O}}_2}\left( {{\rm{core}}} \right).$$
Recently, we systematically analyzed the electrochemical performance of several MnO2 samples with different physical and chemical properties. Reference Zhang, Arthur, Ling and Mizuno32 It was found that the surface area of MnO2 cathode significantly affected the initial discharge capacity. Below 60 m2/g the initial discharge capacity rapidly increased with surface area. Above this value the initial discharge capacity remained nearly constant at about 250 mAh/g. This trend was not affected by the exact polymorphism and chemical composition of MnO2. Therefore the magnesiation process was mainly controlled by the surface of MnO2, consistent with the conclusion that the magnesiation of MnO2 is unlikely to occur via the intercalation of Mg2+.
To understand these experimental results, first-principles calculations were performed to analyze the magnesiation behavior of α-MnO2 by comparing the reactions following different reaction routes. Reference Ling, Zhang, Arthur and Mizuno33 It was found that the conversion reaction that forms amorphous (Mg,Mn)O x is thermodynamically more preferable than the intercalation. Even if the metastable intercalation could occur due to possible kinetic barrier that prevents direct conversion, the concentration of intercalated Mg is limited to α-Mg0.125MnO2, beyond which the structure of intercalated compound undergoes a strong tetragonal to orthorhombic distortion. The thermodynamic driven force for the conversion reaction is attributed to the high stability of MgO. Therefore it was speculated that the conversion reaction may be a general phenomenon for oxide based Mg battery electrodes.
The performance of other MnO2 polymorphs was also studied. Rasul et al. compared the discharge performance of layered-MnO2 and α-MnO2. Reference Rasul, Suzuki, Yamaguchi and Miyayama34 They found that the layered phase showed an initial capacity of 109 mAh/g and α-MnO2 had a very high capacity of 310 mAh/g. The capacity of α-MnO2 was further increased to 475 mAh/g when it was tested at higher temperature, 60 °C. Recently, Rasul et al. also reported the magnesiation performance of MnO2 with even bigger tunnel (octahedral molecular sieves with 4 × 2 tunnel) and got an initial capacity of 78 mAh/g before serious capacity fading. Reference Rasul, Suzuki, Yamaguchi and Miyayama35
Kim et al. compared the electrochemical performance of nanosized λMnO2 and α-MnO2. Reference Kim, Chang, Kim, Kim, Han, Lee, Lee and Doo36 In a three-electrode cell using 0.5 M Mg(ClO4)2 in acetonitrile electrolyte, λMnO2 showed a higher capacity (330 mAh/g) and better cycliability. They believed that it was due to the facile interfacial reaction on the particle surfaces with higher Mg binding energy.
It should be noted that all these reports failed to observe clear voltage plateau in any MnO2 phase. Instead, the voltage profile developed as a sloped curve with large discharge–charge hysteresis. These characters are typically signs of high kinetic battier during the cycling. In addition, the dissolution of Mn3+ species in the electrolyte and structural deformation caused by Jahn–Teller active Mn3+ are common phenomena in Mn based cathode research. Reference Ling and Mizuno37 These results indicate that significant research efforts are still necessary to improve the performance of MnO2 based cathodes.
An unusually polymorph of MgMn2O4 was predicted to have potential as Mg battery owning to the high Mg mobility. Reference Ling and Mizuno38 It has a structure analogue to that of CaFe2O4, with Mg replacing Ca and Mn replacing Fe in the lattice. With one-dimensional triangular-like channel in the structure, the diffusion barrier for Mg migration at the composition of MgMn2O4 was predicted to be ∼0.40 eV, which well matches Li diffusion for many Li-ion battery cathodes. Besides, the voltage to remove Mg from this phase is predicted to contain several plateaus, all of which lie within the stability window of current electrolytes. If the full capacity of this compound can be utilized, the energy density of this cathode is ∼1.3 times to current Li-ion battery cathode.
Unfortunately, this unusual polymorph of MgMn2O4 is only thermodynamically stable at high pressures. Although it may still be kinetically stable at ambient conditions, the synthesis with sufficient amount is still challenging. Three synthetic routes have been suggested to obtain this compound, all of which require a pressure of at least ∼6 GPa. Reference Ling and Mizuno38
Another oxide compound that has been widely studies as Mg battery cathode is layered V2O5. Jiao et al. tested the performance of V2O5 nanotubes in the Grignard electrolyte of 0.25 M Mg(AlBu2Cl2)2/THF solution. Reference Jiao, Yuan, Wang, Cao and Wang39 The initial capacity was about 80 mAh/g with a slope voltage profile averaged at 0.5 V. Imamura et al. prepared V2O5/carbon composition and obtained a high initial capacity up to 550 mAh/g in a three-electrode cell using 1 M Mg(ClO4)2/acetonitrile electrolyte. Reference Imamura, Miyayama, Hibino and Kudo40 Imamura et al. further studied the structural evolution upon magnesiation/demagnesiation process through x-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR) techniques, which clearly identified the intercalation of Mg2+ ion in the layered structure. Reference Imamura and Miyayama41
Recent work by Aurbach's group on V2O5 significantly advanced our understanding of this cathode material in Mg battery system. Reference Gershinsky, Yoo, Gofer and Aurbach42 They systematically investigated the morphology and structural change of V2O5 thin film in the three-electrode cell using 0.1 M Magnesium bis(trifluoromethanesulfonyl)imide/acetonitrile (Mg(TFSI)2/CAN) or 0.5 M Mg(ClO4)2/ACN as electrolyte and carbon paper as counter electrode. A voltage plateau at 2.3 V was observed with the typical discharge capacity of 150 mAh/g at the current density of 0.5 µA/cm2. As shown in Fig. 4, the capacity of V2O5 thin film cathode was stabilized to at least 35 cycles. The electrochemical intercalation/deintercalation of Mg was highly reversible as proven through cyclic voltammetry, XRD, Raman characterization. Scanning electron microscope (SEM) revealed minor morphology change during the cycling.

Figure 4. (a) Galvanostatic titration curve of V2O5 thin-film electrode. (b) Cycling performance and Columbic efficiency of V2O5 thin-film electrode. Reprinted with permission from Ref. Reference Gershinsky, Yoo, Gofer and Aurbach42. Copyright 2013 American Chemical Society.
Zhou et al. carried out first-principles calculations to elucidate the magnetization mechanism of V2O5 cathode. Reference Zhou, Shi, Cao, Zhang and Jiang43 They predicted the open circuit voltage (OCV) of lithium cell using V2O5 cathode at 2.84 V, while the OCV of Mg cell was 3.06 V. The diffusion barrier of Mg2+ in V2O5, however, was predicted to be significantly higher than that of Li+. For Li+ diffusion, the lowest energy barrier was 0.35 eV along b-axis, while for Mg2+ the barrier was 1.26 eV. This large barrier actually indicated the insertion rate would not be practical. The authors also predicted possible phase transformation during the insertion of Mg2+ ions. They concluded that the slow diffusion and phase transition should be two main challenges when using V2O5 as Mg battery cathode.
Wang et al. considered the usage of single-layered V2O5 as cathode candidate for Mg battery. Reference Wang, Su and Deng44 They predicted the adsorption of Mg ions adsorbs on single-layered V2O5 occurred at a voltage of 2.03 V. The diffusion of Mg, however, was not enhanced even in single-layered V2O5. Instead, the activation energy barriers of Mg diffusion in single-layered and bulk V2O5 were 1.40 and 1.36 eV, respectively. It indicates that Mg mobility cannot be enhanced by decreasing the thickness of V2O5 material.
Some other oxides have also been reported as Mg battery cathode in recent years. Sutto et al. investigated Co3O4 and RuO2 as cathode materials for Mg batteries in ionic liquid electrolytes (MMBITFSI and MMOITFSI) using a two-electrode cell with Mg as counter and reference electrodes. Reference Sutto and Duncan45,Reference Sutto and Duncan46 An initial capacity of 74 mAh/g with a voltage plateau at 1.4 V was observed for Co3O4 cathode. The first discharge capacity of RuO2 reached ∼100 mAh/g with surprisingly high voltage plateau at 3.5 V. However, both cathode materials suffered serious capacity fading. The structures of both oxides were significantly changed after discharge. However the structure of the recharged phase was not analyzed.
Another oxide attracted attention is layered MoO3. Aurbach's group studied the magnesiation property of MoO3 thin film. The experiment was performed in a three-electrode configuration using 0.1 M MgTFSI2/AN as electrolyte and porous carbon as counter electrode. Reference Gershinsky, Yoo, Gofer and Aurbach42 A voltage plateau at 1.7 V with the capacity of about 220 mAh/g was observed. Interestingly, the voltage hysteresis of MoO3 was much higher than that of V2O5 thin film (0.4 V versus 0.1 V), which was attributed to the lower diffusivity of Mg2+ in MoO3 and relatively bigger MoO3 particle size.
Spinel oxides are one of the biggest families of cathodes that have been studied in Li-ion battery. Unlike layered LiCoO2, the spinel compounds offer three-dimensional network of interstitial sites for Li diffusion. In Mg battery research, Ichitsubo et al. reported the study of Co and Ni spinel oxides as the potential high voltage cathode materials. Reference Ichitsudo, Adachi, Yagi and Doi47 The performance of MgCo2O4 and Mg0.67Ni1.33O2 were tested in two-electrode cell using Mg metal as anode and 1 M Mg(ClO4)2 in acetonitrile solution as electrolyte. Both showed relative high OCV (∼3.5 V). However, the rate capability was extremely low due to the sluggish Mg2+ diffusion in the host materials. For instance, the authors estimated it took about 300 h to charge 4% of the theoretical capacity of MgCo2O4 (260 mAh/g). Nevertheless, those Co and Ni spinel oxides can be promising cathode materials for magnesium battery providing that the Mg2+ diffusivity can be significantly improved and novel electrolyte can be developed to sustain the high working potential without passivation problem.
To find oxides suitable as Mg battery cathodes, Liu et al. performed first-principles calculation for a series of spinel compounds MgA2O4 with A = Ti to Ni. Reference Liu, Rong, Malik, Canepa, Jain, Ceder and Persson48 All these compounds exhibit lower voltage than that of Li analogues. It is mainly a result of the high redox potential of Mg/Mg2+. However, the bivalency of Mg2+ ions still makes it promising as high energy density cathode. For instance, the capacity of LiMn2O4 is 143 mAh/g, whereas the gravimetric capacity of MgMn2O4 is ∼270 mAh/g, which compensate the sacrifice of the lower voltage.
The diffusion of Mg2+ ions was examined at high vacancy and dilute vacancy limitations, which were believed to set the bound for the diffusion. They found that the migration barrier at high vacancy limit is always higher compared to the dilute vacancy limit. For Li migration in M2O4 (M = Mn, Co, Ni, Cr), the barriers all lied within ∼400 to 600 meV in the empty lattice limit. However, Mg2+ has the barrier about ∼200 to 400 meV higher. It suggested that the diffusivity of Mg might be 4–5 orders of magnitude slower than Li in the spinel phase. Thus the sluggish diffusion of Mg2+ remains as the biggest challenge to use spinel cathode. The best candidate seems to be spinel MnO2, which has a barrier ∼650 meV. Reference Liu, Rong, Malik, Canepa, Jain, Ceder and Persson48
Consistent with Liu et al.'s report, Kim et al. reported the observation of reversible Mg intercalation into spinel-MnO2 using aqueous Mg(NO3)2 eletrolyte (1 M) and Pt as the counter electrode. Reference Kim, Philips, Key, Yi, Nordlund, Yu, Bayliss, Han, He, Zhang, Burrell, Klie and Cabana49 The discharge delivered a capacity of 190 mAh/g. Using a combination of analysis techniques such as scanning transmission electron microscopy (STEM)/energy dispersive x-ray analysis (EDX) and XRD, they found the results were consistent with the formation of MgMn2O4, which was distinct with the amorphourization that was observed in the magnesiation of α-MnO2. It should be noted that the spinel-MnO2 was prepared by acidic leaching of lithium from spinel-LiMn2O4, which also generated nanoflake-like morphology potentially benefit to improve Mg diffusion with shorter migration distance.
Polyanion cathodes
Besides the oxide family, another classical type of intercalation cathodes is polyanion compound. Compared with oxides, the induction effect of the polyanion groups moves the oxygen p-orbital to deeper level, thus generally provides better chemical stability of the cathodes. Research for polyanion compounds as Mg battery cathode mostly used 3d transition metal (V to Ni) as redox center to balance the charge neutrality when guest Mg is inserted or removed.
The most prominent polyanion cathode in Li-ion battery is olivine LiFePO4, which was first reported by Goodenough in 1997. Reference Padhi, Nanjundaswamy and Goodenough50 After that other LiMPO4 olivine compounds with M stands for Mn, Fe, Co, and Ni or their combinations have been extensively studied in Li-ion battery. In LiMPO4 M exists as M2+ ions. If Li is replaced by Mg and phosphorus is replaced by silicon, it ends as MgMSiO4 where M remains as M2+. In fact, the name of the mineral olivine comes from MgMnSiO4. Fully extracting Mg from MgMSiO4 ends at MSiO4, where M exists as 4+. It gives a theoretical capacity of ∼308 mAh/g.
Nuli and her co-workers have systematically examined the performance of Mg olivine cathodes. The performance of these compounds as Mg battery cathode was first reported for Mg1.03Mn0.97SiO4. Reference Feng, Yang, NuLi, Wang, Wang and Wang51 Here the slight off-stoichiometry comes from the anti-site defects formed by switching the position of Mg and transition metal M ions, which is a common phenomenon in olivine compounds. The performance of Mg1.03Mn0.97SiO4 cathode was examined using a CR2016 coin cell with Mg-strip counter electrode and Mg(AlCl2EtBu)2/THF (0.25 M) electrolyte. Figure 5 showed the discharge profile for Mg1.03Mn0.97SiO4. Mg1.03Mn0.97SiO4 prepared by solid-state reaction only delivered a capacity of 43.2 mAh/g at the current density of 6.3 mA/g. After ball-milling it with acetylene black, its capacity was improved to 64.5 mAh/g with discharge voltage plateaus at 1.6 and 1.1 V. For the sol–gel derived Mg1.03Mn0.97SiO4/C electrode, the discharge capacities were further improved to 244, 126, and 76 mAh/g at current densities of 3.1, 6.3, and 15.7 mA/g, respectively. This increased capacity was contributed to the smaller particle size of the active material.

Figure 5. (a) Discharge profile of Mg1.03Mn0.97SiO4 and Mg1.03Mn0.97SiO4/carbon cathode synthesized with solid state method. (b) Improved performance of Mg1.03Mn0.97SiO4/C cathode prepared with sol–gel process. Reprinted with permission from Ref. Reference Feng, Yang, NuLi, Wang, Wang and Wang51. Copyright 2008 Elsevier.
Improved discharge capacities of 200 mAh/g (0.3 C rate) and 150 mAh/g (C/5 rate) were achieved using mesoporous Mg1.03Mn0.97SiO4. Reference NuLi, Yang, Li and Wang52 This performance improvement was attributed to the high surface area of the active mass. Nuli et al. also synthesized multiwalled carbon nanotubes/Mg1.03Mn0.97SiO4 composite hierarchical nanostructure, which showed a steady and stable discharge capacity of 100 mAh/g (at C/2 rate) up to 30 cycle. Reference NuLi, Yang, Wang and Li53
The performances of MgFeSiO4, MgCoSiO4, and MgNiSiO4 were also examined. MgFeSiO4 exhibited a steady discharge capacity ∼125 mAh/g at C/10 rate and retain 91.4% of its initial capacity at 20th cycles. Reference Li, NuLi, Yang, Yilinuer and Wang54 Micrometer sized MgNiSiO4 olivine compound exhibited discharge capacity of 121 mAh/g at a rate of 10 mA/g. Bulk MgCoSiO4 showed a small discharge capacity of 70.2 mAh/g at 0.1 C. Reference Sun55 If hierarchically porous MgCoSiO4 architecture was used, MgCoSiO4 delivered a discharge capacity 80 mAh/g (1 C rate) at the 20th cycle. Reference NuLi, Zheng, Wang, Yang and Wang56
Besides olivine compounds, several other polyanion compounds were also reported as Mg battery cathodes. With the aim to improve Mg diffusion, Huang et al. synthesized a stoichiometric fluoro-phosphate, MgFePO4F (MFPF). Reference Huang, Masese, Orikasa, Mori, Minat, Tassel, Kobayashi, Kageyama and Uchimoto57 The monoclinic structure of MFPF (space group: I2/a) is isostructural to both triplite and wagnerite phases, which contains three-dimensional framework for Mg diffusion. When testing in a three-electrode configuration, the average working potentials of MFPF were measured at ∼2.6 V versus Mg/Mg2+ in Mg-ion cell and 3.1 V versus Li/Li+ in Li-ion cell. However, the capacity of MFPF was not high. The reversible capacity was ∼35 mAh/g at C/20 rate.
Orikasa et al. reported a metal-stable phase of MgFeSiO4. Reference Orikasa, Masese, Koyama, Mori, Hattori, Yamamoto, Okado, Huang, Minato, Tassel, Kim, Kobayashi, Abe, Kageyama and Uchimoto58 The synthesis of metal-stable MgFeSiO4 included the electrochemical extraction of 2Li from Li2FeSiO4 followed by the electrochemical Mg insertion. The discharge processes in Mg electrolyte deliver a capacity of approximately 330 mAh/g. The average charge–discharge potential was 2.4 V. The energy density was estimated to be 746 W h/kg. Orikasa et al. also tested the performance of a magnesium battery using MgFeSiO4 and Mg metal as the cathode and anode, respectively, and Mg(TFSI)2–triglyme as the electrolyte. A reversible charge–discharge capacity of 166 mAh/g was obtained. Only half of the theoretical capacity of the cathode material could be obtained which was contributed to the high polarization effect.
The formal valence of the transition metal ions in olivine MgMSiO4 is +2. Upon the full loss of Mg2+ ions, transition metal ions are oxidized to +4. First-principles calculations based on density functional theory (DFT) revealed that the insertion of magnesium occurs via two steps, each of which is characterized by a unique redox couple. Reference Ling, Banerjee, Song, Zhang and Matsui59 In the first stage, from MgMSiO4 to Mg0.5MSiO4, the redox couple is M2+/M3+. In the second stage, from Mg0.5MSiO4 to MSiO4, the redox couple is M3+/M4+.Therefore the redox reactions are written as
 $$0.5{\rm{M}}{{\rm{g}}^{2 + }} + {{\rm{M}}^{4 + }}{\rm{Si}}{{\rm{O}}_4} + {{\rm{e}}^ - } \to {\rm{M}}{{\rm{g}}_{0.5}}{{\rm{M}}^{3 + }}{\rm{Si}}{{\rm{O}}_4},$$
$$0.5{\rm{M}}{{\rm{g}}^{2 + }} + {{\rm{M}}^{4 + }}{\rm{Si}}{{\rm{O}}_4} + {{\rm{e}}^ - } \to {\rm{M}}{{\rm{g}}_{0.5}}{{\rm{M}}^{3 + }}{\rm{Si}}{{\rm{O}}_4},$$
 $$0.5{\rm{M}}{{\rm{g}}^{2 + }} + {\rm{M}}{{\rm{g}}_{0.5}}{{\rm{M}}^{3 + }}{\rm{Si}}{{\rm{O}}_4} + {{\rm{e}}^ - } \to {\rm{Mg}}{{\rm{M}}^{2 + }}{\rm{Si}}{{\rm{O}}_4}.$$
$$0.5{\rm{M}}{{\rm{g}}^{2 + }} + {\rm{M}}{{\rm{g}}_{0.5}}{{\rm{M}}^{3 + }}{\rm{Si}}{{\rm{O}}_4} + {{\rm{e}}^ - } \to {\rm{Mg}}{{\rm{M}}^{2 + }}{\rm{Si}}{{\rm{O}}_4}.$$
The calculated voltages for Li and Mg insertion into olivine compounds are shown in Fig. 6. All the magnesiation voltages were 0.7–0.9 V lower than those of the lithiation. This difference mainly originates from the anodic potential difference (0.67 V) with additional contribution from the different stability of Mg and Li in the same cathode lattice. Except for Fe2+/Fe3+, the calculated voltages were 3–4 V for M2+/M3+ redox couples and about 4 V for M3+/M4+ redox couples. Note the voltages predicted for Mg insertion were much higher than the experimental value from Nuli et al.'s reports. This discrepancy was expected to come from side reactions during the charge–discharge process.

Figure 6. Voltage calculated for the lithiation and magnesiation of olivine compounds. Reprinted with permission from Ref. Reference Ling, Banerjee, Song, Zhang and Matsui59. Copyright 2012 Royal Society of Chemistry.
Because the crucial challenge in the research of Mg battery cathode is to improve the mobility of Mg2+ ions, theoretical calculations can be extremely helpful to predict candidates with high Mg2+ mobility. Wu et al. predicted the performance of tavorite-Mg0.5FeSO4F as Mg battery cathode. Reference Wu, Gao, Wu, Liu, Yang, Zhou and Wang60 The calculated voltage for Mg extraction from Mg0.5FeSO4F is about 2.52 V. This value was about 1.1 V lower than that of lithiation (3.62 V). The insertion of 1 Mg reduced two FeO4F2 units instead of reducing only one FeO4F2 unit, similar to that revealed for olivine compounds. The energy barrier for Mg-vacancy diffusion along the [010] direction is 0.36 eV, much lower than that of many bulk materials. An estimated diffusion coefficient using the calculated activation energy is on the order of 10−9 cm2/s. These results suggest Mg0.5FeSO4F can serve as a promising potential candidate for magnesium battery cathode materials.
Wu et al. predicted the performance of MgVPO4F as Mg battery cathode. Reference Wu, Gao, Wu, Liu, Yang, Zhou and Wang61 The structure of MgVPO4F was adopted to be isostructural to LiVPO4F. The theoretical capacity of MgVPO4F is about 312 mAh/g. The average voltage to extract Mg was predicted to have two plateaus at 2.6 and 1.5 V, corresponding to the redox couples of V4+/V3+ and V3+/V2+, respectively. The diffusion barriers were calculated along [100], [010], [101], and [111] directions in MgVPO4F. The lowest energy barrier for Mg ion diffusion along [111] direction is 0.704 eV. The activation energies for hops in other direction are at least 700 meV higher, suggesting MgVOPO4F is indeed a 1D Mg conductor. The diffusion coefficient is estimated in the order of 10−14 cm2/s.
Transiton metal dichalcogenide
Transition metal dichalcogenides (MX2, M = transition metal and X = S, Se, and Te) such as TiS2 and VS2 were among the first generation of Li-ion battery cathodes dated back to late 1970s. A few reports tested the performance of these compounds in Mg battery cells. Chen and his group investigated the performance of highly exfoliated graphene-like MoS2 (G-MoS2) composed single- and several-layer of highly exfoliated MoS2. Reference Liang, Feng, Yang, Ma, Liang and Chen62 To enhance the kinetics of the anode, Mg nanoparticle (N-Mg) anode was also prepared. The as-prepared samples were spread into Cu (cathode) or Al (anode) foam and the electrochemical test was carried out using CR2032 coin-type cells with a THF solution of Mg(AlCl3Bu)2 as the electrolyte. The performance of different cathode [G-MoS2 and bulk MoS2 (B-MoS2)] and anode (N-Mg and bulk-Mg) combinations were shown in Fig. 7. All cells exhibited average operating voltages around 1.8 V except for the B-MoS2/B-Mg cell, which worked at a slightly lower discharge voltage of 1.64 V. The best performing G-MoS2/N-Mg cell exhibited a discharge capacity of 170 mAh/g. Subsequent discharge–charge cycling tests revealed excellent capacity retention of the G-MoS2/N-Mg cell. After 50 cycles, 95% of the initial capacity was maintained.
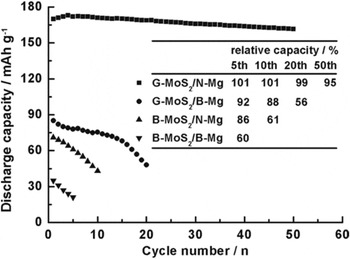
Figure 7. Cycling behaviors of the cells fabricated with B- or G-MoS2 cathode and B- or N-Mg anode with a discharge rate of 20 mA/g. Reprinted with permission from Ref. Reference Liang, Feng, Yang, Ma, Liang and Chen62. Copyright 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Chen et al. carried out a first-principles DFT study focusing on key issues relating to magnesium adsorption sites, theoretical capacity, and diffusion kinetics. Reference Yang, Li, Zhang, Tao and Chen63 In their simulation, MoS2 was treated as zigzag nanoribbons. A maximum theoretical capacity of 223.2 mAh/g was achieved by double-side Mg adsorptions. It was noted that the observed experimental capacity was ∼170 mAh/g. The difference was contributed to possible different crystal structures of MoS2 host in the experiments.
The Mg diffusion pathway on the zigzag MoS2 nanoribbon was identified as passing two adjacent top sites mediated by the nearest neighboring hollow site. The activation barrier of this process was 0.48 eV, much smaller than the barrier in bulk MoS2, 2.61 eV. This theoretical result combined with the experimental observation emphasized the necessity of rational morphology control of electrode materials to enhance the performance of rechargeable Mg electrodes.
The performance of nanostructured MoS2 in Mg battery was reported by Li and Li. Reference Li and Li64 The as-prepared samples was fabricated as cathode in nickel form matrix, and the electrochemical test was conducted using THF solution of Mg(AlCl3Bu)2 as the electrolyte and Mg as the anode. The performance varied cell by cell in the experiments. It was believed that the different sizes and nanostructures of MoS2 caused the different features of the Mg2+ intercalation. The MoS2 nanorods and fullerene-like nanoparticles had small particle size and relative large amount of broken tips, which might be suitable for ions intercalation. Moreover, the interlayer distance of MoS2 fullerene-like nanoparticles were slightly expanded because of the tension. This might also make it easy for the intercalation of Mg2+ ions. For comparison, the commercial MoS2 powder with an average particle size of about 400 nm showed nearly zero capacity for Mg insertion.
Because the diffusion of cations is strongly affected by the distance between slabs, a general strategy to enhance Mg diffusion is to expand the interlayer distance of layered cathode. Reference Doe, Downie, Fischer, Lane, Morgan, Nevin, Ceder, Persson and Eaglesham65,Reference Liang, Yoo, Li, Shuai, Calderon, Hernandez, Grabow and Yao66 With this approach, Liang et al. successfully improved the performance of MoS2 by expanding the interlayer distance from 0.62 to 1.45 nm with controlled pre-insertion of poly(ethylene oxide). Reference Liang, Yoo, Li, Shuai, Calderon, Hernandez, Grabow and Yao66 The average Mg diffusivity over the entire intercalation process measured with galvanostatic intermittent titration technique is 4.4 × 10−12 cm2/s for expanded MoS2, while for in commercial MoS2 the diffusivity is in the order of 10−13 cm2/s once the intercalation begins. Such two orders-of-magnitude boost of Mg diffusion greatly improved the electrochemical performance of MoS2 cathode. The voltage profile for the expanded MoS2 at different current densities is shown in Fig. 8. The capacity at low current density was 75 mAh/g. Even at current density as high as 500 mA/g, the expanded MoS2 still kept a capacity of ∼20 mAh/g. Remarkably, the expanded MoS2 also showed good cyclability at least to 30 cycles, as well as good Columbic efficiency especially at high current densities.
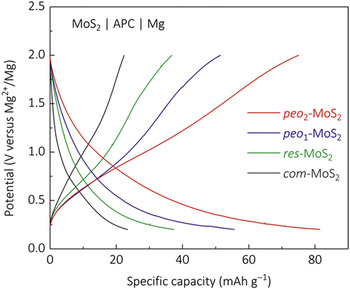
Figure 8. Performance of MoS2 expanded by the insertion of poly(ethylene oxide) in Mg battery. Reprinted with permission from Ref. Reference Liang, Yoo, Li, Shuai, Calderon, Hernandez, Grabow and Yao66. Copyright 2015 American Chemical Society.
Another transition metal dichalcogenide that was tested in Mg cell was WSe2. Reference Liu, Luo, Mu, Wang, Chen and Shen67 The electrochemical properties of WSe2 nanowires with diameters of ∼100 nm were evaluated in coin-shaped cells. The discharge platform was approximately 1.6 V with very little decay of specific capacity during cycling. The reversible specific capacity was around 203 mAh/g. The rate performance of the WSe2 nanowire-based electrode was measured at different current densities ranging from 100 to 800 mA/g for 50 cycles between 0.3 and 3 V. Even at a high current density of 800 mA/g, the specific capacity is still kept at 142 mAh/g. These results indicated the very good structural stability of the WSe2 nanowire-based electrodes.
Oxygen (air) cathode
Motivated by recent progress on Li-oxygen battery, Shiga et al. studied the direct usage of oxygen as cathode for Mg–oxygen battery. Reference Shiga, Hase, Kato, Inoue and Takechi68,Reference Shiga, Hase, Yagi, Takahashi and Takechi69 Ideally the reaction between Mg and O2 occurs via a four-electron transfer in organic solvents as
 $$2{\rm{M}}{{\rm{g}}^{2 + }} + {{\rm{O}}_2} + 4{{\rm{e}}^ - } \to 2{\rm{MgO}}{\rm{.}}$$
$$2{\rm{M}}{{\rm{g}}^{2 + }} + {{\rm{O}}_2} + 4{{\rm{e}}^ - } \to 2{\rm{MgO}}{\rm{.}}$$
The theoretical voltage for this reaction is 3.124 V. Thus the Mg–O2 battery has the potential to deliver energy levels of 1000 (W h)/L if the cathode delivers a capacity more than 320 (mA g)/cc. Unfortunately, the conductivity of the product, magnesium oxide, is very low, while its stability is very high thermodynamically and electrochemically. At ambient temperatures, MgO formed at the O2-electrode cannot be decomposed. Reference Shiga, Hase, Kato, Inoue and Takechi68
To make a rechargeable Mg–O2 battery, Shiga et al. proposed to add redox active species into the electrolyte solution to enhance the dissolution and decomposition of MgO. One of such additives is iodine, which forms complex with organic solvent by a charge-transfer interaction between the functional group in the solvent and the s bond of the iodine molecule. Reference Shiga, Hase, Kato, Inoue and Takechi68 Figure 8 shows discharge–charge curves of a Mg–O2 battery at 60 °C with a carbon electrode containing MgI2 in 0.5 M Mg(ClO4)2–Dimethyl sulfoxide (DMSO). The battery showed two steps in the cell voltage during the first discharging, corresponding to the reaction of Mg2+ with iodine at ∼1.5 V and the discharge of Mg–O2 battery at ∼1.25 V. The total discharge capacity was 2131 mAh/g cathode. In the charge, a plateau was observed at about 2.2 V with a total charge capacity was 1590 mAh/g. If no iodine was added in the electrolyte, the Mg–O2 battery had a large discharge capacity but no charging activity.
Shiga et al. also reported the usage of the 2,2,6,6-tetramethylpiperidine-oxyl–anion complex to catalyze the decomposition of MgO in the charge of Mg–oxygen battery. Reference Shiga, Hase, Yagi, Takahashi and Takechi69 Figure 9 shows the discharge–charge voltage profile for Mg–oxygen battery with a poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl methacrylate) (PTMA) cathode at 60 °C. The discharge capacity of the first cycle was 737 mAh/g while the initial cycle charging capacity was 460 mAh/g. In the second cycle, the discharging capacity was 399 mAh/g, and the charging capacity was as high as 175 mAh/g. The Mg–O2 battery exhibited rechargeable behavior over several discharge/charge cycles.

Figure 9. Discharge–charge profiles of the non-aqueous Mg–O2 battery with iodine at 60 °C. Reprinted with permission from Ref. Reference Shiga, Hase, Kato, Inoue and Takechi68. Copyright 2013 Royal Society of Chemistry.
Sulfur cathode
Another oxygen family element, sulfur, also reacts with magnesium. The theoretical capacity of sulfur is 1671 mAh/g or 3459 mAh/cc. The combination of a magnesium anode and a sulfur cathode is of great interest because the theoretical energy density of this battery is estimated to be over 4000 W h/L, which is approximately twice that of a Li ion battery composed of a graphite anode and a cobalt oxide cathode. It indicates Mg–S battery can serve as a high energy storage system. Reference Kim, Arthur, Allred, Zajicek, Newman, Rodnyansky, Oliver, Boggess and Muldoon7
Compared to the substantial progress made on Li/S batteries, the Mg/S battery is still in a very early stage of research and development. The biggest challenge to realize a rechargeable Mg–S cell is to avoid the direct reaction between the electrophilic sulfur cathode and the nucleophilic electrolyte. Reference Kim, Arthur, Allred, Zajicek, Newman, Rodnyansky, Oliver, Boggess and Muldoon7 Therefore, the development of Mg–S battery heavily relies on the research of novel electrolyte. The first proof-of-concept of Mg–S battery was reported by Muldoon et al. at Toyota, using a metallic magnesium anode, a sulfur cathode dispersed in carbon black, combined with a nonnucleophilic electrolyte based on a recrystallized HMDS–AlCl3 in a coin cell configuration. Reference Kim, Arthur, Allred, Zajicek, Newman, Rodnyansky, Oliver, Boggess and Muldoon7 A typical Mg/S coin cell displayed a capacity of 1200 mAh/g for the first discharge. The starting potential is 0.55 V and slowly increases up to 0.89 V during the discharge process. The second discharge capacity dramatically decreased to 394 mAh/g. The severe capacity fading was attributed to the polysulfide or sulfur dissolution, which also explains the overcharging behavior observed in Fig. 10.

Figure 10. Discharge and charge profiles of Mg/S coin cell. Reprinted with permission from Ref. Reference Kim, Arthur, Allred, Zajicek, Newman, Rodnyansky, Oliver, Boggess and Muldoon7. Copyright 2011 Nature Publishing Group.
To improve the performance of Mg–S battery Zhao-Karger et al. used (HMDS)2Mg based diglyme and tetraglyme electrolyte solutions to build Mg/S batteries and N-methyl-N-butylpiperidinium bis(trifluoromethanesulfonyl)imide (PP14TFSI) as an additive to the electrolytes. Reference Zhao-Karger, Zhao, Wang, Diemant, Behm and Fichtner70 The active sulfur cathode used a highly ordered mesoporous carbon (CMK-3) for the encapsulation of sulfur. The highest discharge capacity was recorded around 800 mAh/g in the first cycle and decreased to 350 mAh/g in the second cycle. After more than 20 cycles it remained a reversible capacity of about 260 mAh/g.
Based on their data, Zhao-Karger et al. proposed that the discharge process of Mg–S battery is divided in three reductive steps. Reference Zhao-Karger, Zhao, Wang, Diemant, Behm and Fichtner70 The first step is a solid–liquid two-phase reduction from elemental sulfur to MgS8, followed by subsequently dissolving of MgS8 into electrolyte to become a liquid cathode and transforms to low-order polysulfides (MgS4). The overall electrochemical reaction for the first step of Mg/S battery could be described as
 $${{\rm{S}}_8} + 4{{\rm{e}}^ - } + 2{\rm{M}}{{\rm{g}}^{2 + }} \to 2{\rm{Mg}}{{\rm{S}}_4}.$$
$${{\rm{S}}_8} + 4{{\rm{e}}^ - } + 2{\rm{M}}{{\rm{g}}^{2 + }} \to 2{\rm{Mg}}{{\rm{S}}_4}.$$
The second step is a liquid–solid two-phase reduction from the dissolved low-order polysulfide to MgS2 corresponding to the second discharge plateau, which results in a theoretical capacity of the Mg/S cell up to 840 mAh/g and can be described as
 $${\rm{Mg}}{{\rm{S}}_4} + 2{{\rm{e}}^ - } + {\rm{M}}{{\rm{g}}^{2 + }} \to 2{\rm{Mg}}{{\rm{S}}_2}.$$
$${\rm{Mg}}{{\rm{S}}_4} + 2{{\rm{e}}^ - } + {\rm{M}}{{\rm{g}}^{2 + }} \to 2{\rm{Mg}}{{\rm{S}}_2}.$$
The third discharging step is a reduction from MgS2 to MgS expressed as
 $${\rm{Mg}}{{\rm{S}}_2} + 2{{\rm{e}}^ - } + {\rm{M}}{{\rm{g}}^{2 + }} \to 2{\rm{MgS}}.$$
$${\rm{Mg}}{{\rm{S}}_2} + 2{{\rm{e}}^ - } + {\rm{M}}{{\rm{g}}^{2 + }} \to 2{\rm{MgS}}.$$
The final product, MgS, is insoluble in ethereal solvents and the process shown in Eq. (7) consequently suffers from high kinetic barriers and high polarization. The latter stages of the conversion reactions in Mg/S batteries proceed sluggishly and limits the initial discharge capacity of about 800 mAh/g, corresponding to the electrochemical reduction of S8 to MgS2.
Mg–anion battery
Due to the low diffusivity of Mg2+ ions in solid state lattice, an alternative solution to secure practically high rate capability is to utilize another redox reaction on the cathode side that replaces the intercalation reaction and balances the electron transfer. This actually is a novel concept for rechargeable Mg battery reported by our group, where the charge transfer is achieved via simultaneous transport of Mg2+ cations and halide anions during electrochemical cycling and the charge balance derived from Mg2+ and X− is compensated in the electrolyte media. We have successfully demonstrated the high rate capability of this conceptual Mg battery using AgCl as cathode material and Cl− containing Grignard electrolyte [Fig. 11(a)]. Reference Zhang, Ling and Mizuno71 The electrochemical reaction during the operation of this battery is the decomposition and formation of metal chloride on the cathode, together with the dissolution and deposition of metallic Mg on the anode. The electrochemical reactions can be described as follows:
 $${\rm{Cathode}}:\quad {\rm{AgCl}} + {{\rm{e}}^ - } \leftrightarrow {\rm{C}}{{\rm{l}}^ - } + {\rm{Ag}},$$
$${\rm{Cathode}}:\quad {\rm{AgCl}} + {{\rm{e}}^ - } \leftrightarrow {\rm{C}}{{\rm{l}}^ - } + {\rm{Ag}},$$
 $${\rm{Anode}}:\quad 0.5{\rm{Mg}} \leftrightarrow 0.5{\rm{M}}{{\rm{g}}^{2 + }} + {{\rm{e}}^ - },$$
$${\rm{Anode}}:\quad 0.5{\rm{Mg}} \leftrightarrow 0.5{\rm{M}}{{\rm{g}}^{2 + }} + {{\rm{e}}^ - },$$
 $${\rm{Electrolyte}}:\quad 0.5{\rm{M}}{{\rm{g}}^{2 + }} + {\rm{C}}{{\rm{l}}^ - } \leftrightarrow 0.5{\rm{MgC}}{{\rm{l}}_2}{\rm{.}}$$
$${\rm{Electrolyte}}:\quad 0.5{\rm{M}}{{\rm{g}}^{2 + }} + {\rm{C}}{{\rm{l}}^ - } \leftrightarrow 0.5{\rm{MgC}}{{\rm{l}}_2}{\rm{.}}$$

Figure 11. (a) Discharge–charge voltage profiles of Mg/AgCl battery at constant current of 0.1 mA. (b) Cycling performance of this cell at different C rates. The inset shows the voltage profiles at 5 C rate. Reprinted with permission from Ref. Reference Zhang, Ling and Mizuno71. Copyright 2015 Royal Society of Chemistry.
Figure 11(b) shows the discharge profiles at different C rates. The theoretical capacity of this cathode is 186 mAh/g. This value was almost achieved at 0.1 C rate. The capacity observed at 10 C still remains 104 mAh/g (55% of the theoretical capacity).
Fitchner's group reported the usage of Mg anode in a halide ion battery cell. Reference Zhao, Li, Zhao-Karger, Gao, Fink, Shen and Fichtner72 They applied metal oxychlorides (BiOCl, FeOCl) as cathode materials, Mg/C composition as anode and a mixture of 0.5 M PP14Cl in PP14TFSI as electrolyte. The BiOCl cathode had an initial discharge capacity of 102 mAh/g, which is 99% the theoretical capacity (103 mAh/g). A charge capacity of 69 mAh/g was recovered in the first cycle, showing a Columbic efficiency of 68%.
Mg–cation battery
Another hybrid ion configuration is Mg–cation cell, where the anode reaction utilizes the electrochemical deposition/dissolution of Mg and the cathode reaction is the insertion of another cation species. Cheng et al. applied Mg metal, mixture of All-Phenyl Complex (APC) and LiCl in THF solution and CP-Mo6S8 as anode, electrolyte and cathode, respectively. Reference Cheng, Shao, Zhang, Sprenkle, Liu and Li73 Due to the difference of diffusivity of Li+ and Mg2+ in CP, Li+ ion intercalates into cathode ahead of Mg2+ ion. On the other hand, the relative higher standard potential of Mg2+ allows Mg metal deposits on anode side. Figure 12 shows typical charge–discharge profiles of a hybrid cell operated at different C-rates. The hybrid cells had two discharge plateaus at 1.66 and 1.29 V and reached a specific capacity of 126 mAh/g at 0.1 C. The capacity had a slight decrease with increase of the C-rate, but still maintained 105 mAh/g at 15 C (83% retention).
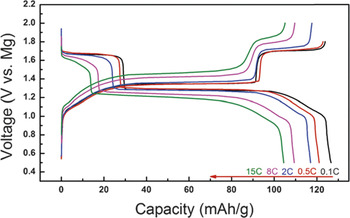
Figure 12. Charge–discharge profiles of the Mg–Li hybrid cell at different C-rates with CP cathode. Reprinted with permission from Ref. Reference Cheng, Shao, Zhang, Sprenkle, Liu and Li73. Copyright 2014 Royal Society of Chemistry.
Cho et al. investigated the reaction mechanism of Mg–Li hybrid battery both experimentally and theoretically. Reference Cho, Aykol, Kim, Ha, Wolverton, Chung, Kim and Cho74 Using DFT calculations, they found that the intercalation of Mo6S8 cathode can be tuned by varying the activity of Li+ ions in the electrolyte. With high Li+ activity the cathode reaction prefers the insertion of Li instead of Mg. By controlling the insertion chemistry using a Li–Mg ions dual-salt electrolyte, they achieved ultrafast discharge at 20 C with 93.6% of capacity retention and at 30 C with 87.5% of capacity retention, respectively, at room temperature.
Similar Mg–Li hybrid battery was also explored by Yagi et al. They used metal Mg as the anode, LiFePO4 as the cathode and THF solution of two kinds of salts, LiBF4 and phenylmagnesium chloride (PhMgCl) as the electrolyte. Reference Yagi, Ichitsubo, Shirai, Yanai, Doi, Murase and Matsubara75 The discharge capacity at the 1st cycle was about 124 mAh/g while the 1st charge capacity was 170 mAh/g in the hybrid electrolyte. The discharge capacity in 2nd cycle dropped to about 96 mAh/g, which was attributed to possible electrolyte decomposition during charge in addition to the self discharge. Especially, the possible reaction between BF4 and PhMgCl would generate B(Ph)4, which might lower the stability of the electrolyte.
It is now recognized that the design of Mg–Li hybrid cell request special attention to the choice of hybrid electrolyte and Li cathode. The presence of lithium salt in the Mg complex electrolyte directly affects its electrochemical properties. The chosen cathode must be compatible with the complex electrolyte, i.e., it should have redox potential within the electrochemical stability window of the complex electrolyte and also remains chemically inert to electrolyte components to avoid any parasitic reactions. On the basis of these considerations, Gao et al. used TiS2 as the cathode and APC–LiCl in THF as the electrolyte in their experiments. Reference Gao, Han, Zhu, Suo, Luo, Xu and Wang76 TiS2 cathode barely delivers any capacity in pure Mg cell. Reference Amir, Vestfrid, Chusid, Gofer and Aurbach77 In Mg–Li hybrid cell, stable electrochemical chemical activity was recorded as shown in Fig. 13. Despite a low initial Columbic efficiency of 90% due to the trapping of inserted Li between TiS2 layers, the discharge–charge curve showed highly reversible performance. No capacity decay was observed for at least 400 cycles with Columbic efficiency as high as 99.5% except for the first few cycles. The theoretical specific capacity and specific energy of the mixed-ion battery are 161.0 mAh/g and 209.3 W h/kg taking into account the weight of lithium salt.

Figure 13. Voltage profile of Mg/PhMgCl–AlCl3–LiCl/TiS2 battery. Reprinted with permission from Ref. Reference Gao, Han, Zhu, Suo, Luo, Xu and Wang76. Copyright 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Prussian blue based cathode
Because of the cost-friendly synthesis and the good rate performance, Prussian blue and its analogues recently have attracted much attention as a potential choice of grid scale storage electrodes. Research in this field focuses on Li and Na battery, with little attention paid to Mg battery. Theoretically it was predicted that the magnesiation of iron hexacyanoferrate, Fe(Fe[CN]6), occurs with two voltage plateaus at 1.93 and 1.40 V, respectively. Reference Ling, Chen and Mizuno78 The maximum capacity is ∼200 mAh/g, corresponds to the insertion/extraction of 1 Mg per formula. Thus the energy density of this compound is not comparative to current Li-ion battery cathode. Nonetheless, the low cost and potentially high rate performance still indicate it may have potential in large scale energy storage.
Cui and his group have extensively studied the performance of hexacyanoferrates in aqueous solutions. Recently they reported the performance of nickel hexacyanonferrate (NiHCF) as divalent ion battery electrode in aqueous electrolyte solutions. Reference Wang, Wessells, Huggins and Cui79 The half-charge reaction potentials versus standard hydrogen electrode (SHE) were 0.60 V (Fig. 14). There was a distinct second plateau in the potential profile at high C rates, suggesting the possibility of a two-step insertion reaction. From C/5 to 10 C rate the retention of specific capacity of 63% was recorded. Besides, only marginal voltage hysteresis was observed at high cycling rate. These results suggested excellent rate performance of NiHCF for Mg (de)intercalation.

Figure 14. (a) Discharge–charge voltage profile of NiHCF upon galvanostatic cycling with Mg2+. (b) Cycling profile of this cell at 5 C rate. Reprinted with permission from Ref. Reference Wang, Wessells, Huggins and Cui79. Copyright 2013 American Chemical Society.
NiHCF also exhibited excellent cycling stability. When cycled between 0.22 and 0.92 V versus SHE at 5 C it retained 65% of initial capacity after 2000 cycles. Round-trip energy efficiency reached 82% after 2000 cycles. If a small concentration of additional Ni2+ (20 mM) was added in the electrolyte to prevent the dissolution of the active material, the energy efficiency was improved to 93%. The capability to react with divalent insertion cations at high rates with unprecedented cycle life and efficiency is essentially interesting for the usage in large scale applications.
Mizuno et al. examined the performance of K0.1Cu[Fe(CN)6]0.7·3.6H2O in a 1 M Mg(NO3)2 aqueous electrolyte. Reference Mizuno, Okubo, Hosono, Kudo, Oh-ishi, Okazawa, Kojima, Kurono, Nishimura and Yamada80 A specific capacity of approximately 60 mAh/g was reversibly obtained for both cathodic and anodic processes, corresponding to 0.3 Mg2+ intercalation/deintercalation per formula unit if this capacity is assumed to entirely come from Mg2+ (de)intercalation. The redox process involved the participation of both Fe and Cu ions with the voltage profile containing a considerable slope over the entire voltage range. At the specific current of 0.1 and 1 A/g, the capacity dropped to 50 and 37 mAh/g, respectively. The retained capacity at 1 A/g reached 74% of the initial maximum capacity, suggesting high rate capability of this material in aqueous solutions.
An interesting question that remains for future research is the intercalation mechanism in aqueous solutions. Reference Wang, Wessells, Huggins and Cui79,Reference Mizuno, Okubo, Hosono, Kudo, Oh-ishi, Okazawa, Kojima, Kurono, Nishimura and Yamada80 The ionic size of Mg2+ is quite small compared to typical ions (K+, Ba2+, etc.) that stabilize the hexacyanonferrate framework. However, the intercell bottleneck radius of the hard sphere model of Prussian blue analogues is about 1.6 Å, while the size of hydrated Mg2+(H2O)6 is about 2.1 Å. Therefore the hydrated Mg2+ may undergo partially dehydration at the interface before the intercalation. This interface process is strongly affected by the choice of solvent. Therefore replacing water with nonaqueous electrolytes may have great impact on the rate performance of Prussian blue electrodes.
Summary and perspective
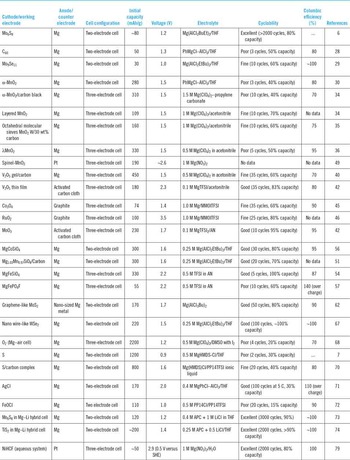
Despite the promising potential of Mg battery as high energy density storage media, the research of Mg battery is still in the infant stage even one and half decade after the Aurbach's Nature report. Reference Aurbach, Lu, Schechter, Gofer, Gizbar, Turgeman, Cohen, Moskovich and Levi6 One of the biggest challenges in Mg battery research is the apparent lack of appropriate cathode that could marry Mg anode in a practical electrochemical device. Reference Yoo, Shterenberg, Gofer, Gershinsky, Pour and Aurbach3,Reference Huie, Bock, Takeuchi, Marschilok and Takeuchi81 Table 2 summarizes the performance of reported cathodes and testing condition that have been reviewed above. Although a series of compounds have been reported to be active as Mg battery cathode, there is no candidate that fully satisfies the requirement for high energy density, good recyclability and nice stability. Significant breakthroughs are still necessary to push forward the activities in this field.
Table 2. Summary of the performance of cathodes in Mg battery.
To discuss the perspective of Mg battery research, let us first look at the energy density of Mg battery. Because of the anode potential of Mg/Mg2+ is higher than that of Li/Li+, it is commonly accepted that the voltage of Mg cell should be typically lower than the Li counterpart by as much as 1.0 V or higher. Therefore, the energy density of Mg battery cathode can only be comparable to Li battery cathode if the former has greater capacity. However, if we consider the energy density of a battery, instead of the energy density of the cathode, the weight or volume of other components should also be taken into account. From this point of view, the larger volumetric capacity of metal Mg anode as well as its nondendritic deposition characteristics stands out as the biggest advantage of Mg battery. If we consider the whole volume of the anode and the cathode, the energy density of the active material is estimated as
 $$U = {{\left( {{V_{{\rm{cathode}}}} - {V_{{\rm{anode}}}}} \right){C_{{\rm{cathode}}}}} \over {1 + {{{C_{{\rm{cathode}}}}} \mathord{\left/ {\vphantom {{{C_{{\rm{cathode}}}}} {{C_{{\rm{anode}}}}}}} \right. \kern-\nulldelimiterspace} {{C_{{\rm{anode}}}}}}}}.$$
$$U = {{\left( {{V_{{\rm{cathode}}}} - {V_{{\rm{anode}}}}} \right){C_{{\rm{cathode}}}}} \over {1 + {{{C_{{\rm{cathode}}}}} \mathord{\left/ {\vphantom {{{C_{{\rm{cathode}}}}} {{C_{{\rm{anode}}}}}}} \right. \kern-\nulldelimiterspace} {{C_{{\rm{anode}}}}}}}}.$$
Figure 15 shows the estimated energy density for several representative configurations. Clearly Mg battery has good potential to reach the energy density of current Li-ion technology using LiCoO2 and graphite as the electrodes. A cell voltage slightly higher than 2.5 V is already competitive to LiCoO2/graphite technology even for low capacity cathode. If we pursue more competitive technology such as LiCoO2/Li electrode, it seems that one may have two directions to improve the energy density. The first direction is to look for high voltage cathode using redox couples such as Ni3+/Ni4+ or Co3+/Co4+. In this way it is possible to increase the voltage of the battery up to 4 V. However, it must be kept in mind that the increase of voltage also requires the development of electrolyte with better stability against oxidation. At current moment the state-of-art electrolyte for Mg battery only provides stability windows up to 3.5 V. Reference Muldoon, Bucur, Oliver, Sugimoto, Matsui, Kim, Allred, Zajicek and Kotani14 Therefore the exploration of high voltage cathode still eagerly requests the breakthrough from the electrolyte study to make the electrochemical test possible.

Figure 15. Theoretical energy densities of Mg battery using cathodes with different capacities.
Another direction is to increase the capacity of the cathode via accomplishing the transfer of two-electron per transition metal ion. In principle, if we can achieve the two-electrode transfer, the capacity can be roughly increased from ∼150 to ∼300 mAh/g. In this case the voltage of Mg battery cathode is only required to be 2 V to match the energy density of LiCoO2/Li battery. This value stays well within the stability window of current electrolyte.
To achieve two-electron transfer, the oxidation state of transition metal ions must be reduced by two. Candidates of redox couples include M6+/M4+, M5+/M3+, and M4+/M2+. 3d transition metal ions typically can be reduced from 4+ to 2+. However, based on first-principles calculations, it seems that direct reduction from 4+ to 2+ may not be practical for most 3d transition metal ions. For instance, the extraction of Mg from olivine MgMnSiO4 occurs via the intermediate M3+ state. The redox potential of M4+/M3+ and M3+/M2+ can vary by 1 V in Li-ion battery. Reference Hautier, Jain, Mueller, Moore, Ong and Ceder82 Therefore, in Mg battery test it is likely that either the voltage of M3+/M2+ becomes too low for a cathode, or M4+/M3+ becomes too high for the electrolyte. Besides, the reduction from M4+ to M2+ may greatly change the unit cell volume by >10%, Reference Ling, Banerjee, Song, Zhang and Matsui59 which challenges the structural integrity of the cathode. These facts indicate the M4+/M2+ may not be a practical choice for a two-electron transfer cathode. On the other hand, certain transition metal ions such as Mo6+/Mo4+ and W6+/W4+ favor direct two-electron reduction. Cathode utilizing these redox couples may be more interesting as Mg battery cathode. However, up to our knowledge the study based on these redox couples is not systematically carried out except for MoO3. Reference Gershinsky, Yoo, Gofer and Aurbach42
The second and perhaps the biggest challenge for the research of Mg battery cathode is related to the kinetic performance. More specifically, because of the bivalent Mg2+ interacts stronger with the host ions, the diffusion of Mg2+ is more sluggish than the monovalent Li+. This behavior has two direct impacts. First, it leads to very poor rate performance of an intercalation cathode, or even prevents the (de)intercalation of Mg2+ with a practical rate. Second, the slow diffusion generates a large kinetic barrier, which results in large discharge–charge voltage hysteresis. This damages the efficiency of the battery to store the energy.
Because of the complicated nature about the diffusion of Mg2+ in a solid state material, the fundamental understanding at current moment is still very limited. Simulations based on first-principles calculations can be particularly helpful because it can reveal the detailed information about Mg diffusion in the atomic level. However, calculation about the diffusion is more computational expensive than those geometry optimizations. Therefore at current moment the Mg diffusion is only analyzed for selected compounds. Reference Ling and Mizuno38,Reference Zhou, Shi, Cao, Zhang and Jiang43,Reference Wang, Su and Deng44,Reference Liu, Rong, Malik, Canepa, Jain, Ceder and Persson48,Reference Huang, Masese, Orikasa, Mori, Minat, Tassel, Kobayashi, Kageyama and Uchimoto57,Reference Wu, Gao, Wu, Liu, Yang, Zhou and Wang60,Reference Wu, Gao, Wu, Liu, Yang, Zhou and Wang61,Reference Yang, Li, Zhang, Tao and Chen63 A few compounds have been proposed with high Mg mobility including post-spinel MgMn2O4, Reference Ling and Mizuno38 spinel MgMn2O4, Reference Liu, Rong, Malik, Canepa, Jain, Ceder and Persson48 MoS2, Reference Yang, Li, Zhang, Tao and Chen63 and tavorite-Mg0.5FeSO4F, Reference Wu, Gao, Wu, Liu, Yang, Zhou and Wang60 all of which have diffusion barrier less than 700 meV. Except for MoS2, the experimental validation about the electrochemical performance is still requested. Nonetheless, considering the large configuration space of possible Mg containing compounds, we are optimistic that large scale screening can identify candidates with better Mg mobility in future.
Because the major hurdle for the low kinetic performance of Mg cathode is the sluggish diffusion of Mg2+ ions in the cathode host, one strategy to avoid it is to use a hybrid ion battery. Typical hybrid ion battery includes Mg anode, a mixture of Mg2+ and other ions in the electrolyte and a cathode. During the cycling, the anode still experiences the deposition and dissolution of Mg, while the cathode reaction does not necessarily require the participation of Mg2+. A configuration like that still keeps the advantage of Mg anode without suffering from the slow Mg intercalation kinetics. In sections “Mg–anion battery” and “Mg–cation battery,” the hybrid Mg–anion and Mg–cation battery both showed potentially high rate capability. Unlike the classical Li-ion battery where the electrolyte only participates as the shuttle of Li+ ions, in the hybrid ion battery the electrolyte is involved into the reaction and thus affects the cell voltage. The Nernst equation gives the voltage of Mg–anion battery as
 $$V = kT\,{\rm{log}}\left[ {{{\left( {{{{a_{{\rm{Mg}}}}} \over {{a_{{\rm{M}}{{\rm{g}}^{2 + }}}}}}} \right)}^{0.5}}{{\left( {{{{a_{\rm{X}}}} \over {{a_{{{\rm{X}}^{n - }}}}}}} \right)}^{1/n}}} \right].$$
$$V = kT\,{\rm{log}}\left[ {{{\left( {{{{a_{{\rm{Mg}}}}} \over {{a_{{\rm{M}}{{\rm{g}}^{2 + }}}}}}} \right)}^{0.5}}{{\left( {{{{a_{\rm{X}}}} \over {{a_{{{\rm{X}}^{n - }}}}}}} \right)}^{1/n}}} \right].$$
For Mg–cation battery, the voltage becomes
 $$V = kT\,{\rm{log}}\left[ {{{\left( {{{{a_{{\rm{Mg}}}}} \over {{a_{{\rm{M}}{{\rm{g}}^{2 + }}}}}}} \right)}^{0.5}}{{\left( {{{{a_{{{\rm{X}}^{n + }}}}} \over {{a_{\rm{X}}}}}} \right)}^{1/n}}} \right].$$
$$V = kT\,{\rm{log}}\left[ {{{\left( {{{{a_{{\rm{Mg}}}}} \over {{a_{{\rm{M}}{{\rm{g}}^{2 + }}}}}}} \right)}^{0.5}}{{\left( {{{{a_{{{\rm{X}}^{n + }}}}} \over {{a_{\rm{X}}}}}} \right)}^{1/n}}} \right].$$
Here a stands for the activity of ions in the electrolyte and in the electrode. The concentration, stability, and the dissociation/association kinetics of the electrolyte thus serve as an important role in the hybrid ion battery. Nonetheless, because it avoids the slow Mg2+ diffusion, hybrid ion battery has the potential to deliver much higher rate capability.
Finally, we should point out again that the research of Mg battery cathode should not be isolated from other components of the cell. Otherwise the limited knowledge of Mg battery chemistry may be misleading to interpret the experimental data. It is now recognized that the commonly used Mg battery electrolyte, the Grignard reagent, is corrosive to many current collectors. Reference Muldoon, Bucur, Oliver, Sugimoto, Matsui, Kim, Allred, Zajicek and Kotani14,Reference Muldoon, Bucur, Oliver, Zajicek, Allred and Boggess15 If the electrochemical test was carried out using nonresistant current collectors, the corrosion might be mistakenly interpreted as the activity of the cathode occurring at low voltages (usually around 2 V). To avoid such misinterpretation, careful analysis must be taken to reveal the real reaction on the cathode. On the basis of our experience, we suggest that several techniques should be applied, including applying XPS to analyze the reversible valence change of the active redox species, XRD, SEM, and TEM to explore the structure evolution during the cycling, and element analysis (EDS and inductively coupled plasma) to demonstrate the incorporation of Mg into the cathode. More advanced technique such as synchrotron XAS analysis can be extremely helpful from this aspect.
CONCLUSION
Great advances have been achieved in the past few years in Mg battery research. However, the lack of appropriate cathode still badly prevents this technology from any practical application. In the current review, we provide a comprehensive analysis about the performances of Mg battery cathode reported in the past 15 years, focusing on the voltage, capacity, rate performance, and cyclability data. We clearly see that none of the reported cathode satisfies the requirement of high energy density, good rate capability, and nice cyclability to compete with current Li-ion technology. However, we believe this should not be treated as a discouragement for Mg battery. In fact, a simple analysis reveals that Mg battery has good potential to compete with Li-ion battery that uses graphite anode, or even with future Li-ion battery that probably use metal Li as the anode. Three important areas that must be explored in this field in near future are suggested: the investigation of high capacity cathode, the study of hybrid ion battery, and the deeper understanding about the magnesiation chemistry of the cathode. We therefore hope this review could act as a benchmark for future Mg battery cathode research.

















