


Maternal obesity predisposes offspring to metabolic disorders. Nutritional overload during early or prenatal life can cause permanent damage to offspring( Reference Danielzik, Langnäse and Mast 1 – Reference Catalano, Farrell and Thomas 3 ). Consumption of a high-fat diet (HFD) during pregnancy leads to the activation of macrophages in the maternal placenta that culminates in an inflammatory environment for fetal development( Reference Challier, Basu and Bintein 4 ). Maternal body-weight gain during critical phases of offspring development results in greater adiposity, body weight, liver TAG content and hepatic steatosis in adult life( Reference Elahi, Cagampang and Mukhtar 5 , Reference Ashino, Saito and Souza 6 ). Furthermore, the uterus and blastocysts from obese dams exhibit inflammatory signals linking maternal obesity to increased predisposition of offspring to obesity later in life( Reference Shankar, Zhong and Kang 7 ).
Liver steatosis is characteristic of obesity and diabetes and is closely associated with inflammatory signals( Reference Qureshi and Abrams 8 , Reference Stanton, Chen and Jackson 9 ). Maternal consumption of a HFD during pregnancy has been shown to result in increased fetal hepatic lipid accumulation, oxidative stress and apoptosis in a non-human primate model of maternal obesity( Reference McCurdy, Bishop and Williams 10 ). Recently, we observed that offspring of obese dams displayed increased fatty liver deposition immediately after weaning and into their adulthood( Reference Ashino, Saito and Souza 6 ). In addition, Heerwagen et al. ( Reference Heerwagen, Stewart and de la Houssaye 11 ) showed that, at embryonic day 18.5, fetuses of obese dams had higher fetal liver TAG deposition than those of lean dams. The metabolic pathways leading to the development of hepatic steatosis are multiple, including enhanced NEFA release from adipose tissue (lipolysis), increased de novo fatty acid synthesis (lipogenesis) and decreased β-oxidation activity( Reference Postic and Girard 12 , Reference Bouanane, Merzouk and Benkalfat 13 ).
Epigenetic mechanisms have been associated with metabolic imprinting of and damages to adulthood offspring( Reference Waterland 14 , Reference Sullivan and Grove 15 ). MicroRNA (miRNA) are endogenous non-coding RNA of approximately twenty-two nucleotides in length that regulate various metabolic processes and diseases( Reference Bartel 16 ). miRNA-122 (miR-122) is liver-specific and the most abundant miRNA in this organ, accounting for approximately 70 % of the total miRNA population. Functional studies have shown that miR-122 is involved in multiple metabolic processes including cholesterol biosynthesis, fatty acid synthesis and oxidation( Reference Li, Xi and Zhu 17 ). The repression of miR-122 results in hepatic insulin resistance by protein-tyrosine phosphatase 1B induction( Reference Yang, Seo and Kim 18 ), similar to the repression of miRNA-370 (miR-370). Iliopoulos et al. ( Reference Iliopoulos, Drosatos and Hiyama 19 ) showed that miR-370 directly down-regulated the expression of the gene encoding carnitine palmitoyltransferase 1α (CPT1α), that controls fatty acid oxidation. Therefore, miRNA may contribute to the induction of metabolic damage associated with fatty liver deposition in the offspring of obese dams. We used offspring mice that were recently weaned from obese dams to investigate the modulation of hepatic fatty acid synthesis (de novo), β-oxidation pathways, and expression of miR-122 and miR-370 by maternal consumption of a HFD during pregnancy and lactation.
Materials and methods
Ethics statement
All experiments were performed in accordance with the guidelines of the Brazilian College for Animal Experimentation (COBEA) and approved by the Committee for Ethics in Animal Experimentation (protocol no. 2864-1) at the State University of Campinas – UNICAMP (Campinas, São Paulo, Brazil).
Animals
A total of ten virgin female and male Swiss mice (7 weeks old) that were specific pathogen free were obtained from the Animal Breeding Center at the University of Campinas (Campinas, SP, Brazil) for mating. Before mating, the females were randomly fed ad libitum either a HFD or standard laboratory chow (SC) for 2 weeks for adaptation (Table 1) and received filtered water ad libitum. Mating was performed by housing females with adult males (fed SC) for 1 week, and pregnancy was confirmed by the examination of vaginal smears for the presence of sperm.
Table 1 Nutritional composition of the experimental and standard chow diets fed to mice during gestation and lactation

* NUVILAB® Cr-1; Nuvital.
Pregnant females were maintained in individual polypropylene microisolators with a bed of pinewood autoclaved (about 60 g) in a rack at 22 ± 1°C with lights on from 06.00 to 18.00 hours. They received the same diet (HFD or SC) during pregnancy and lactation as before mating. The HFD was prepared according to AIN-93G modified for high fat (45 %) content (Table 1). Offspring were divided into two groups according to maternal feeding: offspring of female mice fed the HFD (HFD-O group) and offspring of female mice fed the SC (SC-O group). At day 1 after birth, the litters of both groups (HFD-O and SC-O) were adjusted to eight pups each to ensure a standard litter size per mother. The pups were weaned on day 18 and separated according to sex. Male offspring were maintained in the same environmental conditions fed the SC after weaning until the end of the experimental period (day 28), according to the experimental protocol (Fig. 1).
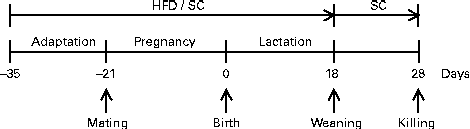
Fig. 1 Experimental protocol followed to obtain offspring of dams fed a high-fat diet (HFD; HFD-O mice) and standard chow (SC; SC-O mice). Difference between the groups is based only on the diet offered to dams during the periods of adaptation, pregnancy and lactation. After weaning, HFD-O and SC-O mice were fed the SC diet.
Sample size was estimated from previous experiments and confirmed using a simple power and sample size calculation, available at the website of the Biomathematics Division of the Department of Pediatrics at the College of Physicians and Surgeons at Columbia University( Reference Dell, Holleran and Ramakrishnan 20 ). The number of individual experiments was representative of at least three different litters.
The total number of animals used in each experiment is indicated in the figure legends.
Biochemical analysis
At the end of the experimental period (day 28) and after overnight fasting, all mice were killed and blood samples were collected and centrifuged, and serum aliquots were used to measure the levels of serum TAG (glycerol-3-phosphate oxidase–phenol+aminophenazone (GPO-PAP); Roche Diagnostics), cholesterol (cholesterol oxidase–phenol+aminophenazone (CHOD-PAP); Roche Diagnostics) and NEFA (acyl-CoA synthase–acyl-CoA oxidase (ACS-ACOD); Wako Chemicals) by enzymatic colorimetry.
Frozen tissues (200 mg) from SC-O and HFD-O specimens were homogenised in 1·5 ml of PBS and processed as described by others( Reference Carr, Andresen and Rudel 21 , Reference Newberry, Xie and Kennedy 22 ). The protein concentration of the homogenate was determined, and an aliquot of 300 μl was extracted with 5 ml of chloroform–methanol (2:1) and 0·5 ml of 0·1 % H2SO4 (v/v). An aliquot of the organic phase was collected, dried under N2 and resuspended in 2 % Triton X-100. TAG content was determined using a commercially available kit (GPO-PAP; Roche Diagnostics).
Intraperitoneal glucose tolerance test and intraperitoneal insulin tolerance test
For intraperitoneal glucose tolerance test, offspring of HFD- and SC-fed mice at day 28 were starved for 12 h, fed for 2 h and starved for an additional 4 h before intraperitoneal injection of glucose (1 g/kg of a 25 % solution of d-glucose) as described by Bonora et al. ( Reference Bonora, Zavaroni and Alpi 23 ). Blood samples were collected from the tail at 0, 10, 15, 30, 60 and 120 min after injection for the measurement of blood glucose concentration. The AUC of glycaemia v. time was calculated above each individual baseline (basal glycaemia) to estimate glucose tolerance. The AUC of glycaemia (mmol/l) v. time (120 min) was calculated above each individual baseline (basal glycaemia) to estimate glucose tolerance, using the trapezoidal method.
For intraperitoneal insulin tolerance test, insulin (6·00 nmol (1·0 IU)/kg body weight) was administered by intraperitoneal injection, and tail blood samples were collected at 0, 3, 6, 9 and 12 min after insulin administration to mice (day 28), following the same fasting protocol. The constant for the glucose disappearance rate during the test( Reference Stenseth, Viljugrein and Saitoh 24 ) was calculated using the formula 0·693/t 1/2 ( Reference Bonora, Zavaroni and Alpi 23 ).
For both glucose tolerance test and insulin tolerance test, glycaemia was determined on an Accu-Chek Performa glucometer (Roche Diagnostics).
Food intake measurement
Food intake was estimated during 24 h over a period of four alternate days after weaning. The average was considered as food intake (g/d).
Quantitative real-time PCR
Total hepatic RNA was extracted using TRIzol reagent (Life Technologies Corporation), according to the manufacturer's recommendations. Total RNA was quantified on a NanoDrop ND-2000 (Thermo Electron) and its integrity verified by agarose gel electrophoresis. Reverse transcription was performed with 3 μg of total RNA and a High-Capacity cDNA Reverse Transcription kit (Life Technologies Corporation). Relative expression was determined using the Taqman detection system and primers for the target genes: Mm 00772290_m1 stearoyl-coenzyme A desaturase 1 (SCD1); Mm 01282499_m1 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR); Mm 01304277_m1 acetyl-CoA carboxylase α (ACACA); Mm 00662313_m1 fatty acid synthase (FASN); Mm 00479699_g1 1-acylglycerol-3-phosphate O-acyltransferase 1 (AGPAT1); Mm 01247712_m1 hepatocyte nuclear factor 4, α (HNF4α); Mm 00444293_m1 acyl-CoA dehydrogenase, very long chain (ACADVL); Mm 01231183_m1 CPT1α; Mm 01296700_m1 AMP-activated protein kinase (AMPK) (Life Technologies Corporation); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the endogenous control (4352339E mouse GAPD; Life Technologies Corporation). Each PCR contained 20 ng of complementary DNA.
Gene expression was quantified by real-time PCR performed on an ABI Prism 7500 Fast platform. Data were analysed using the Sequence Detection System 2.0.5 (Life Technologies Corporation) and expressed as relative values determined by the comparative threshold cycle (C t) method (2− ΔΔC t ), according to the manufacturer's recommendation.
MicroRNA isolation and quantification
miRNA was extracted and purified from the liver of SC-O and HFD-O mice using a mirVana miRNA Isolation Kit (Life Technologies Corporation). The relative expression of miR-122 and miR-370 was determined using the TaqMan detection system (Life Technologies Corporation), the appropriate primers (ID 002245 and 002275, respectively), and U6 spliceosomal RNA (ID 001973) and miR-16 (ID 000391) as endogenous controls (Life Technologies Corporation). Gene expression was quantified as described in the preceding section.
Immunoblotting
Tissue samples (day 28) were homogenised in freshly prepared ice-cold buffer (1 % (v/v) Triton X-100, 0·1 m-Tris, pH 7·4, 0·1 m-sodium pyrophosphate, 0·1 m-sodium fluoride, 0·01 m-EDTA, 0·01 m-sodium vanadate, 0·002 m-phenylmethanesulfonyl fluoride and 0·01 mg aprotinin/ml). The insoluble material was removed by centrifugation (10 000 g ) for 25 min at 4°C. The protein concentration of the supernatant was determined by the Bradford dye-binding method. The supernatant was resuspended in Laemmli sample buffer and boiled for 5 min before separation by SDS–PAGE using a miniature slab gel apparatus (Bio-Rad). Electrotransfer of proteins from the gel to nitrocellulose was performed for 90 min at 120 V (constant). The nitrocellulose blots were probed with specific antibodies. Antibodies to phospho-c-Jun N-terminal kinase (JNK, SC-1648), phospho-IκB kinase (SC-21 660), NF-κBp65 (SC-8008), fatty acid synthase (FAS, SC-20 140), HMGCR (SC-33 827), CPTI (SC98834), ACADVL (SC-376239) and HNF4α (SC-8987) were obtained from Santa Cruz Biotechnology, Inc. Antibodies against phospho-acetyl-CoA carboxylase (ACC, 3661S), ACC (3676S), phospho-AMPK (2535S) and AMPK (2532S) were obtained from Cell Signaling Technology, Inc. Antibodies against β-actin (ab8227) and SCD1 (ab19862) were obtained from Abcam.
After incubation with specific antibodies, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (KPL). Proteins recognised by the secondary antibodies were detected by chemiluminescence (Amersham ECL kit (RPN 2232)) as visualised by the exposure of the blot to Kodak XAR film. Band intensities were quantified by optical densitometry of developed autoradiographs (Scion Image software; ScionCorp), and the intensities of the bands were normalised to those of total protein or the loading control β-actin.
Liver histochemistry analysis
Fragments of liver from the HFD-O and SC-O mice were fixed in 10 % formalin for 24 h. Tissue samples were subsequently processed using routine histological methods and embedded in paraffin. Briefly, tissues were sectioned (5 μm), stained with haematoxylin–eosin and analysed by light microscopy (Leica FW 4500 B microscope; Leica Imaging Systems). Samples were coded and assessed by two independent, blinded observers.
Statistical analysis
All numerical results are expressed as means with their standard errors of the indicated number of experiments. Blot results are presented as direct band comparisons in autoradiographs and quantified by densitometry using Scion Image software (ScionCorp). Student's t tests of unpaired samples were employed for determining a significance level of P< 0·05.
Results
To examine the effects of maternal HFD-induced obesity on offspring metabolism and miRNA expression, we first characterised our experimental model. Table 2 shows that in early gestational and lactation periods, HFD-fed dams had a higher body weight than SC-fed dams (1·5- and 1·3-fold, respectively). HFD-fed dams also showed higher (17-fold) epigonadal adipose tissue than SC-fed dams. On day 1, the body weights of SC-O and HFD-O mice did not differ significantly. At day 28, HFD-O mice were significantly heavier (approximately 1·3-fold) than SC-O mice. In addition, HFD-O mice had larger epididymal (3·1-fold) and retroperitoneal (2·0-fold) fat pads than SC-O mice (Table 3). Furthermore, food intake was little higher in HFD-O (1·1-fold) than in SC-O mice (Table 3). In agreement with these results, maternal consumption of the HFD during pregnancy and lactation resulted in glucose intolerance and insulin resistance in the offspring, as indicated by the AUC during the glucose tolerance test and κITT (intraperitoneal insulin tolerance test constant) calculated from the insulin tolerance test (Table 4), respectively. In addition, serum components such as cholesterol, TAG and NEFA were more abundant in HFD-O mice than in SC-O mice (Table 4).
Table 2 Body composition and glycaemia from the control (standard chow (SC))- and high-fat diet (HFD)-fed dams (Mean values with their standard errors)
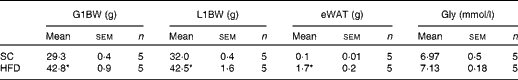
G1BW, body weight at first gestational week; L1BW, body weight at first lactational week; eWAT, epigonadal white adipose tissue at day 28; Gly, fasting glycaemia at day 28.
* Mean value was significantly different from that of the SC-fed group (P≤ 0·05).
Table 3 Body composition and food intake of the offspring of dams fed a standard chow (SC-O group) and high-fat diet (HFD-O group) (Mean values with their standard errors)
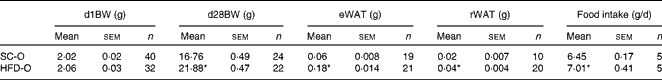
d1BW, body weight at first postnatal day; d28BW, body weight at postnatal day 28; eWAT, epididymal white adipose tissue; rWAT, retroperitoneal white adipose tissue.
* Mean value was significantly different from that of the SC-O group (P≤ 0·05).
Table 4 Metabolic parameters of the offspring of dams fed a standard chow (SC-O group) and high-fat diet (HFD-O group) at day 28 (Mean values with their standard errors; n 6–12 pups per group)
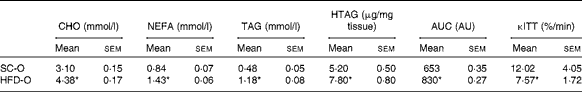
CHO, serum cholesterol; TAG, serum TAG; HTAG, hepatic TAG; AUC, AUC of serum glucose curve during the glucose tolerance test; AU, arbitrary units; κITT, intraperitoneal insulin tolerance test constant for serum glucose disappearance.
* Mean value was significantly different from that of the SC-O group (P≤ 0·05).
Because HFD consumption leads to peripheral inflammation and defective regulation of energy homeostasis( Reference Ashino, Saito and Souza 6 ), pro-inflammatory pathways in the liver were investigated (day 28). As shown in Fig. 2, hepatic IκB kinase phosphorylation, JNK phosphorylation and NF-κBp65 were higher in HFD-O mice than in SC-O mice (4·0-, 4·3- and 5·2-fold, respectively; Fig. 2(a)–(c)).

Fig. 2 Western blotting (WB) of hepatic phospho-IκB kinase (p-IKK) (a), phospho-c-Jun N-terminal kinase (p-JNK) (b) and NF-κB (NF-κBp65) (c) in the offspring of dams fed a high-fat diet (HFD-O group) and standard chow (SC-O group) (day 28). For the control of gel loading in WB, membranes were reblotted with β-actin. Values are means (n 4–7), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the SC-O group (P≤ 0·05).
We next investigated the influence of maternal obesity on lipogenic gene expression in the offspring. HFD-O and SC-O mice expressed similar levels of hepatic phospho-ACC, FAS and HMGCR and ACC and FASN mRNA (Fig. 3(a) and (c)–(e)). However, HFD-O mice expressed more hepatic AGPAT1 mRNA (1·7-fold; Fig. 3(e)), in addition to SCD1 protein and mRNA (2·2- and 3·0-fold, respectively) than SC-O mice (Fig. 3(b) and (e)), even though HMGCR mRNA levels were reduced (Fig. 3(e)).

Fig. 3 Western blotting (WB) of hepatic phospho-acetyl-CoA carboxylase (p-ACC)/ACC (a), stearoyl-CoA desaturase 1 (SCD1) (b), fatty acid synthase (FAS) (c) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) (d) in the offspring of dams fed a high-fat diet (HFD-O group, ■) and standard chow (SC-O group, □) (day 28). For the control of gel loading in WB, membranes were reblotted with β-actin. mRNA levels (quantitative real-time PCR (qRT-PCR)) of hepatic ACC, SCD1, fatty acid synthase (FASN), 1-acylglycerol-3-phosphate O-acyltransferase 1 (AGPAT1) and HMGCR (e) in mice (day 28). For relative gene expression analysis, glyceraldehyde-3-phosphate dehydrogenase was used as the endogenous control. Values are means (n 4 for WB and n 8–12 for qRT-PCR), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the SC-O group (P≤ 0·05).
To evaluate the influence of maternal obesity on β-oxidation-related gene expression in the offspring, AMPK phosphorylation and ACADVL and CPT1 expression were determined. As shown in Fig. 4(a), hepatic AMPK phosphorylation was similar in SC-O and HFD-O mice (day 28). However, levels of ACADVL protein and mRNA and CPT1 mRNA were lower in HFD-O mice than in SC-O mice (75, 30 and 40 %, respectively; Fig. 4(b) and (c)). In contrast, AMPK mRNA levels were higher (1·3-fold) in the liver of HFD-O mice than in that of SC-O mice (Fig. 4(c)). Interestingly, HFD-O mice had an increased hepatic TAG content (1·5-fold) compared with SC-O mice (Table 4). The presence of vacuoles that contained lipids within hepatocytes as shown by haematoxylin–eosin-stained liver sections from the HFD-O mice (Fig. 5(c) and (d)) corroborates molecular results for this group, while the SC-O group had a normal liver structure (Fig. 5(a) and (b)).

Fig. 4 Western blotting (WB) of hepatic phospho-AMP-activated protein kinase (p-AMPK)/AMPK (a) and acyl-CoA dehydrogenase, very long chain (ACADVL) (b) in the offspring of dams fed a high-fat diet (HFD-O group, ■) and standard chow (SC-O group, □) (day 28). For the control of gel loading in WB, membranes were reblotted with β-actin. mRNA levels (quantitative real-time PCR (qRT-PCR)) of hepatic AMPK, ACADVL and carnitine palmitoyltransferase 1 (CPT1) (c). For relative gene expression analysis, glyceraldehyde-3-phosphate dehydrogenase was used as the endogenous control. Values are means (n 4 for WB and n 8 for qRT-PCR), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the SC-O group (P≤ 0·05).

Fig. 5 Photomicrographs of the liver stained with haematoxylin–eosin from mice at day 28 showing vacuoles that contained lipids (arrows). (a, b) Offspring of dams fed a standard chow (original magnification 200 × and 400 × , respectively, n 8). (c, d) Offspring of dams fed a high-fat diet (original magnification 200 × and 400 × , respectively, n 8).
Compared with SC-O mice, HFD-O mice expressed similar levels of hepatic HNF4α protein and mRNA (Fig. 6(a) and (b)), reduced (25 %) levels of hepatic miR-122 (Fig. 6(c)) and increased (3-fold) levels of miR-370 (Fig. 6(d)).

Fig. 6 Western blotting (WB) (a) and mRNA levels (b) of hepatic hepatocyte nuclear factor 4, α (HNF4α). MicroRNA (miRNA) levels of hepatic miRNA-122 (miR-122) (c) and miRNA-370 (miR-370) (d) in the offspring of dams fed a high-fat diet (HFD-O group) and standard chow (SC-O group) (day 28). For the control of gel loading in WB, membranes were reblotted with β-actin. For relative gene expression analysis, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), miRNA-16 (miR-16) and U6 spliceosomal RNA (U6snRNA) were used as the endogenous controls. Values are means (n 4 for WB and n 8–9 for quantitative real-time PCR), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the SC-O group (P≤ 0·05).
Discussion
It has been demonstrated that high-fat diet consumption activates pro-inflammatory pathways, causes endoplasmic reticulum stress, ectopic lipid deposition and insulin resistance, and contributes to other co-morbidities associated with obesity( Reference Shoelson, Lee and Yuan 25 – Reference Velloso and Schwartz 31 ). Moreover, maternal consumption of a HFD during pregnancy and lactation has also been related to metabolic disturbances in adult offspring( Reference Ashino, Saito and Souza 6 , Reference Shankar, Zhong and Kang 7 , Reference McCurdy, Bishop and Williams 10 , Reference Levin 32 ).
Hepatic damage associated with metabolic changes promoted by maternal consumption of a HFD during pregnancy and lactation has been described in offspring at different periods of development( Reference Ashino, Saito and Souza 6 , Reference McCurdy, Bishop and Williams 10 , Reference Heerwagen, Stewart and de la Houssaye 11 , Reference Bouanane, Merzouk and Benkalfat 13 ). Fatty liver is characteristic of obesity and diabetes and is closely associated with inflammatory signals( Reference Qureshi and Abrams 8 , Reference Stanton, Chen and Jackson 9 ). As shown here, HFD-O mice weighed more, had a larger adipose tissue mass than SC-O mice and were more glucose- and insulin-intolerant. Furthermore, HFD-O mice expressed more hepatic NF-κBp65, phospho-JNK and phospho-IκB kinase than SC-O mice, suggesting that liver insulin resistance may be associated with the activation of pro-inflammatory pathways, as described previously( Reference Donath and Shoelson 28 , Reference Barbuio, Milanski and Bertolo 33 , Reference Cintra, Pauli and Araújo 34 ). However, recent conflicting findings question the importance of hepatic inflammation in the development of insulin resistance. Wiedemann et al. ( Reference Wiedemann, Wueest and Item 35 ) showed that adipose tissue inflammation contributed to HFD-induced hepatic insulin resistance, whereas Turner et al. ( Reference Turner, Kowalski and Leslie 36 ) found that consumption of a HFD for 3–4 weeks induced insulin resistance without the evidence of inflammation in the liver, adipose tissue or skeletal muscle. Moreover, they detected adipose tissue inflammation only after 16 weeks of HFD consumption. Interestingly, TAG content and the amount of a diacylglycerol species were increased in the liver after 1 week of HFD consumption, whereas changes in ceramide abundance occurred only after the development of insulin resistance( Reference Turner, Kowalski and Leslie 36 ). Thus, ectopic lipid accumulation appears to correlate with insulin resistance( Reference Kraegen and Cooney 37 , Reference Samuel and Shulman 38 ), but the lipid classes that mediate insulin resistance are unknown( Reference Farese, Zechner and Newgard 39 ). We did not evaluate inflammatory markers in the adipose tissue, but we demonstrated increased adipose tissue mass and hepatic lipid accumulation in HFD-O mice. Furthermore, it is important to point out that although HFD can impair the effect of insulin on glucose production, insulin-stimulated lipid synthesis was not altered in the present study, indicating selective insulin resistance( Reference Brown and Goldstein 40 ). Thus, damage to hepatic glucose homeostasis can occur in the offspring of HFD-fed dams in the absence of changes in lipid synthesis. Interestingly, in a previous study employing the same model as used here, we demonstrated that HFD-O mice (day 82) exhibited liver insulin resistance and JNK and IκB kinase activation in association with elevated TAG content and reduced phosphorylation of AKT and ACC, limiting the steps of de novo lipid synthesis( Reference Ashino, Saito and Souza 6 ). Similarly, as shown here, HFD-O mice (day 28) exhibited insulin resistance and alterations in glucose homeostasis, although ACC phosphorylation was similar in HFD-O and SC-O mice. Furthermore, although hepatic TAG and serum lipid levels were increased in abundance in HFD-O mice, the expression of enzymes involved in fatty acid biosynthesis (ACC and FAS) was not affected by maternal HFD consumption. However, HFD-O mice highly expressed SCD1, which converts stearate (18 : 0) to oleate (18 : 1) and palmitate (16 : 0) to palmitoleate (16 : 1). Previous studies have shown that inhibition of SCD1 expression by SCD1-specific antisense oligonucleotides reduced blood insulin levels, de novo fatty acid synthesis, steatosis and expression of lipogenic genes, and increased fatty acid oxidisation in primary mouse hepatocytes and the expression of genes promoting energy expenditure in the liver and adipose tissue( Reference Jiang, Li and Liu 41 , Reference Yokoyama, Hosoi and Ozawa 42 ). Recently, animal studies indicating a relationship between SCD1 expression and metabolic disorders have been reported( Reference Hodson and Fielding 43 ). Furthermore, HFD-O mice also increased AGPAT1 expression, which catalyses the conversion of lysophosphatidate to phosphatidate, adding an acyl group to the sn-2 position of the glycerol backbone in the TAG synthesis pathway( Reference Takeuchi and Reue 44 ). The role played by AGPAT1 in hepatic steatosis is still poorly understood. However, a recent study has shown that the knockdown of AGPAT1 in hepatocytes isolated from liver-specific knockout mice of the Mir122 locus reduced TAG synthesis, suggesting that AGPAT1 plays a key role in liver TAG accumulation in these mice( Reference Hsu, Wang and Kota 45 ).
Increased de novo fatty acid synthesis (lipogenesis) and decreased β-oxidation activity can lead to hepatic steatosis( Reference Postic and Girard 12 , Reference Bouanane, Merzouk and Benkalfat 13 ). As evidence, investigators have shown that maternal consumption of a HFD affects total cholesterol and LDL-cholesterol levels and brain fatty acid composition( Reference Elahi, Cagampang and Mukhtar 5 , Reference Yu, Bi and Ma 46 ). In support of the literature, HFD-O mice had more levels of serum cholesterol, NEFA and TAG than SC-O mice. It is important to point out that maternal HFD consumption during lactation can contribute to milk composition( Reference Silber, Hachey and Schanler 47 , Reference Priego, Sánchez and García 48 ), and therefore suckling periods could affect serum lipid levels. Although HFD-O mice received the SC diet for 1 week after weaning, the impact of milk composition on the serum lipid profile cannot be overlooked. In physiological conditions, potential sources of fatty acids that contribute to liver TAG deposition include hydrolysis in adipose tissue, dietary uptake and de novo lipogenesis in the liver.
Studies in human subjects and rodents have shown that excessive accumulation of liver TAG mainly results from the overflow of fatty acids generated by lipolysis in insulin-resistant adipose tissue( Reference Lewis, Carpentier and Adeli 49 , Reference Ferré and Foufelle 50 ). Donnelly et al. ( Reference Donnelly, Smith and Schwarzenberg 51 ) showed that steatosis arose from circulating fatty acids in 60 % of patients and from de novo lipogenesis in 25 % of patients. In agreement with the literature, the present results show that HFD-O mice have more adipose tissue and serum NEFA levels than SC-O mice. Furthermore, maternal consumption of a HFD affected SCD1 (increased expression), AGPAT1 (increased expression) and genes related to fatty acid oxidation (reduced expression of ACADVL and CPT1), but did not affect enzymes important for fatty acid synthesis. Altogether, these results suggest that hepatic TAG accumulation in HFD-O mice can also be due to diminished fatty acid oxidation. Moreover, the high SCD1 enzyme activity could lead to increased availability of MUFA for TAG synthesis in HFD-O mice, and the increase in AGPAT1 expression in HFD-O mice reinforces this hypothesis.
Many studies have indicated the importance of miRNA in lipid metabolism and liver physiology and disease( Reference Iliopoulos, Drosatos and Hiyama 19 , Reference Esau, Davis and Murray 52 – Reference Aranda, Madrigal-Matute and Rotllan 56 ), but the role of these small non-coding RNA in hepatic lipid metabolism is still controversial. Esau et al. ( Reference Esau, Davis and Murray 52 ) showed that inhibition of miR-122 expression reduced fatty acid synthesis and increased fatty acid oxidation. Recently, two studies( Reference Hsu, Wang and Kota 45 , Reference Tsai, Hsu and Hsu 57 ) employing genetic deletion in mice have provided substantial evidence of miR-122 function in lipid metabolism. Hsu et al. ( Reference Hsu, Wang and Kota 45 ) showed the up-regulation of genes involved in lipid synthesis in miR-deficient liver such as AGPAT, phosphatidic acid phosphatase type 2A (Ppap2a) and monoacylglycerol transferases (Mogat). Consistent with the changes in gene expression, the mutant liver synthesised more, but secreted less, TAG than the control liver, resulting in TAG accumulation in mutant hepatocytes. In addition, both groups also showed an increase in the number of infiltrating inflammatory cells in the liver of miR-122-deficient mice( Reference Hsu, Wang and Kota 45 , Reference Tsai, Hsu and Hsu 57 ), suggesting a pro-inflammatory effect of reduced expression of this miRNA. Interestingly, HFD-O mice exhibited reduced expression of miR-122, increased liver TAG deposition and JNK activation, although SCD1 increased in expression. Others have shown that suppression of miR-122 in non-human primates reduces plasma cholesterol( Reference Krützfeldt, Rajewsky and Braich 58 , Reference Elmén, Lindow and Schütz 59 ). In the present study, HFD-O mice expressed less HMGCR than SC-O mice, but the level of plasma cholesterol were increased rather than reduced. It is possible that maternal consumption of a HFD modifies milk composition and affects the cholesterol level in plasma. We also found that HFD-O mice expressed more miR-370 in the liver than SC-O mice. This miRNA controls the expression of miR-122 and affects lipid metabolism( Reference Iliopoulos, Drosatos and Hiyama 19 ). miR-370 activates lipogenic genes indirectly through miR-122 in a different experimental model. Importantly, miR-370 directly down-regulates CPT1α, which controls the rate-limiting step in fatty acid β-oxidation. In agreement with the literature, HFD-O mice also had reduced expression of CPT1α and possibly reduced fatty acid β-oxidation, as indicated by the diminished expression level of ACADVL protein and mRNA.
Thus, hepatic lipid accumulation in the offspring of obese dams has multifactorial characteristics, and changes in the oxidative pathway are associated with the differential expression of miR-370 and miR-122 in the liver. In addition, HFD-O mice also had increased liver inflammation, probably because of the uterine environment. This adaptive response leads to hepatic metabolic changes that cause liver injury.
Acknowledgements
The present study was supported by São Paulo Research Foundation (FAPESP) (grant no. 2011/22156-7) and the National Council for Scientific and Technological Development (CNPq, grant no. 479017/2011-5). The funders had no direct influence over the design, conduct or reporting of the present study.
R. O. B. was supported by São Paulo Research Foundation (FAPESP) Fellow Masters in nutrition science, sport and metabolism (grant no. 2011/13947-0).
R. O. B. contributed to the development of the study hypothesis, reviewed the literature, conducted the statistical analysis, provided interpretation of the results, and drafted and edited the manuscript. A. M. M., F. O. B. and L. A. P. S. contributed to the development of the study hypothesis and provided advice regarding interpretation of the results. L. M. I.-S., M. M. and L. A. V. assisted with drafting and editing of the manuscript. M. A. T. provided advice regarding interpretation of the results and edited the manuscript. A. S. T. was responsible for the study design, data collection, provided advice regarding interpretation of the results and edited the manuscript.
None of the authors has any conflict of interest to declare.










