

Introduction
αIIbβ3 (GPIIb/IIIa) antagonists are effective in decreasing mortality and morbidity when administered to patients undergoing percutaneous coronary artery interventions (PCI), with significant reductions in the odds of both all-cause mortality at 30 days [odds ratio (OR) 0.79, 95% confidence interval (CI) 0.64–0.97)] and death or myocardial infarction at 30 days (OR 0.66, 95% CI 0.60–0.72) in more than 40 studies conducted on more than 30,000 patients [Reference Bosch, Marrugat and Sanchis1]. Whereas thrombi causing ST segment elevation myocardial infarction (STEMI) have in general, but not uniformly, been found to be platelet rich early after symptom onset and less platelet rich over time [Reference Silvain2–Reference Yunoki4], there is reason to expect that potent antiplatelet agents would be more effective when administered early after the onset of symptoms. Consistent with this hypothesis, meta-analyses of studies comparing administration of αIIbβ3 antagonists relatively early after symptom onset to administration at the time of the PCI demonstrated significant benefits from early therapy in pre-PCI blood flow in the target vessel, time to ST segment resolution, left-ventricular ejection fraction, distal embolization, and mortality [Reference De Luca5–Reference Xu, Yin and Si7]. Similar benefits of early versus late abciximab were observed in studies of patients with STEMI transferred for primary PCI who received abciximab before transfer rather than during PCI, with a higher percentage of patent arteries pre-PCI and lower rates of death and a composite clinical outcome at 30-day follow-up [Reference Dudek8]. Most importantly, in the randomized studies, the mortality benefit from early αIIbβ3 antagonist therapy increased over an 11-year period, with those treated within 65 minutes manifesting less than 5% morality at 11 years compared to greater than 30% mortality for those treated >178 minutes after the onset of symptoms [Reference De Luca6].
Despite these data, early treatment of STEMI with an αIIbβ3 antagonist is not in common use, in part because all of the agents require both an intravenous (IV) bolus injection followed by a continuous IV infusion controlled by a pump. We previously reported on a novel small molecule αIIbβ3 antagonist, RUC-4 [Reference Li9], which is a derivative of a compound (RUC-1) we identified in a high throughput screen for inhibitors of platelet adhesion to fibrinogen [Reference Blue10–Reference Zhu12] and RUC-2, which was modified from RUC-1 [Reference Zhu13]. RUC-2 and RUC-4 exclude the divalent cation required for ligand binding from the β3 subunit metal ion-dependent adhesion site (MIDAS) domain and thus prevent ligand binding, αIIbβ3-mediated platelet aggregation, and in vivo thrombus formation [Reference Li9,Reference Zhu13]. Moreover, since the interaction between the ligand’s carboxyl oxygens with the metal ion in the MIDAS triggers the receptor to undergo a major conformational change that places it in a high-affinity ligand binding state [reviewed in [Reference Coller14]], both RUC-2 and RUC-4 lock the receptor in its inactive conformation. This contrasts with other small molecule αIIbβ3 antagonists based on the Arg-Gly-Asp (RGD) sequence, since they act as pseudo-ligands, binding to the MIDAS metal ion through their carboxyl oxygens and inducing the conformational change. RUC-4’s unique mechanism of action may offer clinical advantages since the conformational change produced by the other agents may contribute to the thrombocytopenia observed in a small percentage of treated patients who have preformed antibodies to the altered conformation [Reference Aster15–Reference Brassard17], and induction of the high affinity state may limit efficacy. The latter may even have contributed to the increased risk of death observed with some of the oral αIIbβ3 antagonists, all of which have carboxyl oxygens that are presumed to bind to the MIDAS metal ion [Reference Cox18–Reference Chew, Bhatt and Topol20].
We previously reported that intramuscular administration of RUC-4-free base to male cynomolgus monkeys (Macaca fascicularis) non-human primates (NHPs) at doses between 1 and 3.86 mg/kg led to the onset of high-grade inhibition of platelet aggregation within 15–30 minutes that lasted from ~2 to >4.5 hours in a dose-dependent manner [Reference Li9]. To achieve our goal of making it as easy as possible for first-point-of-medical-contact emergency medical service and emergency room medical personnel to administer RUC-4 rapidly to patients with STEMI, and knowing that it is challenging to reproducibly administer drugs intramuscularly by autoinjector in the emergency setting with a single needle size [Reference Bhalla21,Reference Song22], we have now compared the pharmacokinetics (PK) of RUC-4 administered subcutaneously (SC) with the PK and pharmacodynamics (PD) of RUC-4 administered intramuscularly (IM).
We also conducted additional preclinical studies of RUC-4 using human blood in anticipation of starting human studies. Since immediate aspirin treatment is approved for treatment of myocardial infarction and is considered the current standard of care for STEMI, [Reference Perina and Braithwaite23,Reference O’Gara24] we plan to administer RUC-4 in combination with aspirin to STEMI patients. As a result, we also studied the effect of aspirin on the antiplatelet effect of RUC-4. In addition, since the anticoagulant utilized to prepare platelets for platelet aggregation studies can influence the response to different antiplatelet agents, [Reference Phillips25–Reference Kereiakes28] we also studied the impact of different anticoagulants on the sensitivity of platelets to the inhibitory effects of RUC-4. Finally, to obtain a better understanding of the correlation between inhibition of platelet aggregation and the percentage of receptors blocked by RUC-4, we studied this relationship directly.
Materials and Methods
RUC-4
RUC-4 was synthesized at the National Center for Advancing Translational Science at the National Institutes of Health as previously described or with minor modifications [Reference Jiang29].
Study Comparing the Effects of IM and SC RUC-4 in Cynomolgus Monkey (M. fascicularis) NHPs
Platelet aggregation and Determination of the RUC-4 IC50 in NHPs. Light transmission platelet aggregation studies were performed with a PAP-8E aggregometer (Bio/Data Corporation, Horsham, PA) at 37°C with stirring using 5 µM ADP to induce aggregation. Blood (5–7.5 ml) was obtained from four NHPs with a 21-gauge needle and anticoagulated with 1/10th volume of 3.8% sodium citrate to determine the RUC-4 IC50 for platelet aggregation. Platelet-rich plasma (PRP) was prepared by centrifuging whole blood at 600 × g for 1–2.5 minutes at room temperature. The remaining blood was centrifuged at 1200 × g for 8 minutes at room temperature to prepare platelet-poor plasma (PPP). Platelet counts on PRP were performed with an automated platelet counter (IDEXX ProCyte Dx Hematology Analyzer, Westbrook, ME) and were adjusted down to a count of 3.0 × 105/μl with PPP when the PRP platelet counts were higher. PRP (0.25 ml) was incubated with increasing concentrations of RUC-4 for 20 minutes before initiating aggregation with 5 µM ADP. Platelet aggregation was quantified based on the instrument’s measurement of the primary slope of aggregation and the percentage inhibition produced by RUC-4 calculated by reference to the slope of the untreated sample. To determine the RUC-4 IC50, PRP from four animals was either untreated (control) or incubated with RUC-4 at final concentrations of 0.01, 0.03, 0.1, 0.3, 0.5, 1.0, 3.0, or 5.0 µM before adding ADP at 5 µM. The 0.01 and 5.0 µM doses were only studied in two animals. In one animal, aggregation was not assessed at 1 µM because of technical problems. The percentage inhibition of the initial slope of aggregation relative to the control value was calculated for each concentration for each animal, averaging duplicate values when they were available (11/32 samples), and the RUC-4 IC50 was calculated using the Levenberg–Marquardt algorithm to perform the linear regression fit using the Hill equation.
Animal Handling
This study was performed at Biomere, Worcester, MA, on six adult male animals aged 3.6–4.8 years obtained from Charles River Laboratories. Animals used on study followed the test facility’s standard operating procedures for receipt of NHPs and underwent a physical examination, tuberculosis and measles testing, and evaluation of clinical pathology. All animals were considered in good health by the attending veterinarian before being released for study. The veterinary care of the animals was in accordance with the protocol, Biomere’s standard operating procedures, and regulations outlined in the applicable sections of the Final Rules of the Animal Welfare Act regulations (9 CFR), the Public Health Service Policy on Humane Care and Use of Laboratory Animals, the Guide for the Care and Use of Laboratory Animals, and the Biomedical Research Models, Inc., Policy on Humane Care. The protocol and any amendments or procedures involving the care or use of animals in this study were reviewed and approved by Biomere’s Institutional Animal Care and Use Committee (IACUC) before the initiation of such procedures.
The six NHPs used in these studies were single or socially housed, when possible, in stainless steel cages equipped with a stainless steel mesh floor and an automatic watering valve in a room that was well ventilated and maintained at a temperature of 18–29°C and a relative humidity of 30%–70%. Fluorescent lighting provided illumination approximately 12 hours per day. Animals were fed Monkey Diet 5038 (Lab Diet) daily and filtered tap water was provided ad libitum via an automatic watering system. All animals received environmental enrichment including manipulable devices, foraging opportunities, food items, structural and environmental enhancements, and positive human interaction. Enrichment devices were rotated on a weekly basis and include toys, mirrors, radios, TV, foraging boards, and a variety of complex foraging devices. After the completion of sample collection, the animals were released back to the Biomere colony. Manual and/or mechanical (chair) restraint was used as necessary to facilitate animal handling and performance of the technical procedures. Blood was collected either from a vein or artery in the femoral triangle from animals acclimated to restraint.
Prior to dose administration, hair was shaved and trimmed at the injection site in order to improve the visualization of any injection site observations. RUC-4 was administered as a single IM or IV injection at a constant dose volume of 0.064 ml/kg under manual restraint without any sedation. All IM injections were administered directly into the thigh semitendinous muscle, which was marked for later evaluation. Direct pressure to the injection site was maintained for at least 5 minutes to minimize the risk of bleeding. IV injections were made via an angiocatheter placed into the saphenous vein. The dose volume for each animal was based on the most recent body weight measured on the day before or the day of dosing. SC injections were made on a shaved portion of the abdomen at a constant volume of 0.064 ml/kg using a 23-gauge, ¾ inch needle. The site of injection was compressed manually for 5 minutes after the injection.
Dosing
The six animals were divided into three dose groups of two animals/group and they received RUC-4 at doses of 3.86 mg/kg, 1.93 mg/kg, and 1 mg/kg IM, IV, and SC as indicated in Table 1. Since there were five arms to the study, and each animal was studied in each arm, each animal received repeated doses a total of five times over the course of approximately 6 months.
Table 1. NHP study experimental design
No., number; ROA, route of administration; IV, intravenous; IM, intramuscular; SC, subcutaneous.
The same animals were studied in each arm of the study and so each animal received five doses via different ROAs over a period of 6 months.
Blood Sampling for PK, PD, and Blood Counts
For PK studies, 0.5 ml of whole blood was immediately added to a chilled tube containing 2 ml of a mixture of water and acetonitrile (70:30 vol/vol), and then the tube was immediately vortexed and stored at –80°C until analyzed. In the first four arms, samples were collected and evaluated (n = 2/dose/time point) for PK analysis at pre-dose and at 5, 15, 30, 45, 120 and 270 minutes after IM and IV injection; additional samples were obtained at 1 minute after IV administration and at ~27 hours after administering 3.86 mg/kg IM in arm 1, the latter because the platelet aggregation responses had not returned to baseline levels at 270 minutes. In arm 5, animals (n = 2/dose/time point) received RUC-4 at the same three doses and blood was obtained pre-dose and at 5, 15, 30, 60, and 120 minutes after dosing for PK analysis.
Blood (2.5 ml) for light transmission platelet aggregation studies was collected into 1/10th volume of 3.8% sodium citrate from the animals receiving RUC-4 IM pre-dose and at 30, 120, and 270 minutes after RUC-4; an additional sample was obtained at ~27 hours from animals receiving 3.86 mg/ml RUC-4 in arm 1 because the platelet aggregation responses were still inhibited at 270 minutes post-dose. Blood was centrifuged at 650 × g for 1–2.5 minutes at room temperature to prepare PRP and the remaining blood was centrifuged at 1200 × g for 8 minutes at room temperature to prepare PPP. Platelet counts were performed on PRP as above and samples were adjusted to a count of 3 × 105/μl with PPP if the PRP count was higher.
Blood (0.5 ml) was collected into EDTA for complete blood counts (CBC) in both animals in each dose group in arms 1 and 2 pre-dose and at 30, 120, and 270 minutes post-dose. In arm 3, both animals had blood drawn for a CBC pre-dose and 15 minutes post-dose; in arm 4, both animals had blood drawn for CBC pre-dose and at 45 minutes post-dose; in arm 5, both animals had blood drawn for CBC pre-dose and at 120 minutes post-dose.
Analysis of RUC-4 Whole Blood Concentrations
Whole blood samples collected in water-acetonitrile solution were analyzed for RUC-4 by liquid chromatography with tandem mass spectrometry detection (LC-MS/MS) with an Acquity UPLC BEH HILIC column (130 Å, 1.7 µm; 50 x 2.1 mm) and an API-6500 mass spectrometer in positive ion mode using electrospray and a temperature of 550°C. Mobile phase A was 10 mM ammonium acetate in 100:0.1 (vol/vol) water:NH4OH and mobile phase B was 10 mM ammonium acetate in 100:0.1 (vol/vol) acetonitrile:NH4OH. Purified RUC-4 (387.1/330.1 amu) was used as a calibration standard and deuterated RUC-4 (D8; 395.1/338.1 amu) served as an internal control. The standard curve was based on quadratic regression with acceptance criteria of ± 20% and an accepted curve range of 5–5000 ng/ml. The blood concentrations of RUC-4 after IM and SC administrations were evaluated by non-compartmental analyses (NCA) with Phoenix WinNonlin software (version 6.3, Pharsight, St. Louis, MO, USA).
In Vitro Platelet Aggregation Studies in Healthy Human Volunteers
Blood sampling and preparation of PRP. All studies were approved by the Rockefeller University Institutional Review Board (IRB) and all blood donors gave informed consent. Blood from healthy volunteers was obtained via venipuncture with a 19-gauge needle and anticoagulated with 1/10th volume 3.8% sodium citrate (final concentration 0.38%), ~1/15th volume D-phenylalanyl-prolyl-arginyl chloromethyl ketone (PPACK; final concentration 0.1 or 0.3 mM), or dried sodium heparin in a vacuum tube (Vacutainer, Becton Dickinson) designed to achieve a final concentration of 15.8 USP U/ml). Samples were gently inverted and kept at room temperature until processed. PRP was prepared by centrifuging the whole blood at 650 x g for 4 minutes at room temperature. The remaining blood was centrifuged for 1200 x g for 8 minutes at room temperature to collect PPP. Platelet counts in the PRP were adjusted to 3.0 x 105/µl with PPP. Platelet aggregation studies were conducted by incubating 225 µl of PRP with 2 µl of test compound for 20 minutes at room temperature and then adding 25 µl of agonist with stirring to initiate aggregation.
Preparation of RUC-4 stock solutions for platelet aggregation studies. Working stock dilutions of RUC-4 in arms 1, 2, and 5 were made up in 0.9% sterile saline at concentrations of 375 µM, 125 µM, 62.5 µM, 37.5 µM, 25 µM, 12.5 µM, 3.75 µM, and 1.25 µM to achieve final concentrations of 3 µM, 1 µM, 0.5 µM, 0.3 µM, 0.1 µM, 0.03 µM, and 0.01 µM when 2 µl was added to 250 µl of PRP.
Preparation of agonist stock solutions for platelet aggregation studies. Working stock solutions of a thrombin receptor activating peptide (TRAP; SFLLRN) of 200 µM and ADP of 200 µM and 50 µM were prepared with 0.9% sterile saline and final concentrations 20 and 5 µM were achieved by adding 25 µl to 225 µl PRP. The arachidonic acid stock was made by dissolving 5 mg in 1 ml of water.
Preparation of aspirin-treated PRP. An initial 1 M aspirin stock solution was made by dissolving acetyl salicylic acid in water and a 75 mM working stock solution was made by diluting the initial stock solution with water. The final concentration of aspirin used in the study (0.83 mM) was achieved by adding 2.5 µl of 75 mM aspirin to 225 µl of PRP for 10 minutes, followed by incubation with RUC-4 at the indicated concentrations for 20 minutes before initiating platelet aggregation with either arachidonic acid (1.5 mM) or ADP (5 µM).
Flow cytometry studies with whole blood utilizing monoclonal antibody (mAb) PAC-1 to assess the percentage of receptors occupied by RUC-4. PAC-1 is an IgM mouse mAb that selectively binds to activated αIIbβ3 in the same region occupied by RUC-4 [Reference Shattil30]. As a result, PAC-1 binding to activated platelets identifies the receptors that are not already bound by RUC-4, providing information on the percentage of receptors occupied by RUC-4. In the assay, 25 µl of whole blood from five separate donors (three female and two male) was incubated for 5 minutes at room temperature with 12.5 µl of FITC-labeled 25 µg/ml PAC-1 (final concentration: 6.25 µg/ml), either 0.4 µl of 1 mM PGE-1 (negative control) or 0.4 µl of varying concentrations of RUC-4 (final concentrations 3 µM, 1 µM, 0.5 µM, 0.3 µM, 0.1 µM, 0.03 µM and 0.01 µM), and 27 µl of 1X HBMT with 1 mM Mg2+. Five microliters of 200 µM ADP (final concentration 20 µM) was added to the above mixture and incubated for 30 minutes at room temperature. Four hundred and fifty microliters of 1X HBMT with 1 mM Mg2+ was added to make up the final volume to 500 µl and flow cytometry was conducted immediately at room temperature using the FACSCalibur (Becton Dickinson, BD).
Flow cytometry data were analyzed using BD CellQuest™ Pro Analysis software. Data were expressed as both the geometric mean fluorescence and the percentage of the mean geometric fluorescence obtained in the absence of RUC-4.
Data analysis. Platelet aggregation responses and RUC-4 IC50 determinations were quantified as described previously for the NHP studies.
Results
Studies in NHPs
Determination of RUC-4 IC50 for platelet aggregation in NHP. The RUC-4 IC50 values for the 4 NHPs tested ranged from 499 to 1060 nM, with a mean ± SD of 807 ± 231 nM.
Study comparing the effects of IM and SC RUC-4 in NHPs.
Clinical assessments. IM administration: All animals exhibited some minor bruising and red, raised areas limited to site of injection (thigh) or venipuncture (left and right inguinal areas) shortly after treatment that resolved over time. In addition, minor bruising was seen at sites on the animal where manual restraint was performed (i.e., shoulders and arms). The clinical observations were similar at all doses of RUC-4 tested. On Day 2, clinical observations were for the most part limited to areas of venipuncture with blood sampling, and all animals were considered healthy at the end of the observation period.
IV administration. Animals exhibited similar clinical observations as those seen after IM administration, but one animal in arm 2 and both animals in arm 3 of the study vomited immediately after the IV injection of 3.86 mg/kg. In addition, one animal given 1.93 mg/kg in arm 2 had minor nose and gingival bleeding. On Day 2, clinical observations of bruising were noted as minor or moderate, and limited for the most part to sites of venipuncture and manual restraint.
SC administration. Clinical findings included red discoloration of the skin at the venipuncture and administration sites and bruising at sites of injection and inguinal areas. These clinical signs resolved by ~24 hours post-dose.
All animals were found to be healthy by the examining veterinarian prior to return of the animals to the colony at the end of all arms of the study.
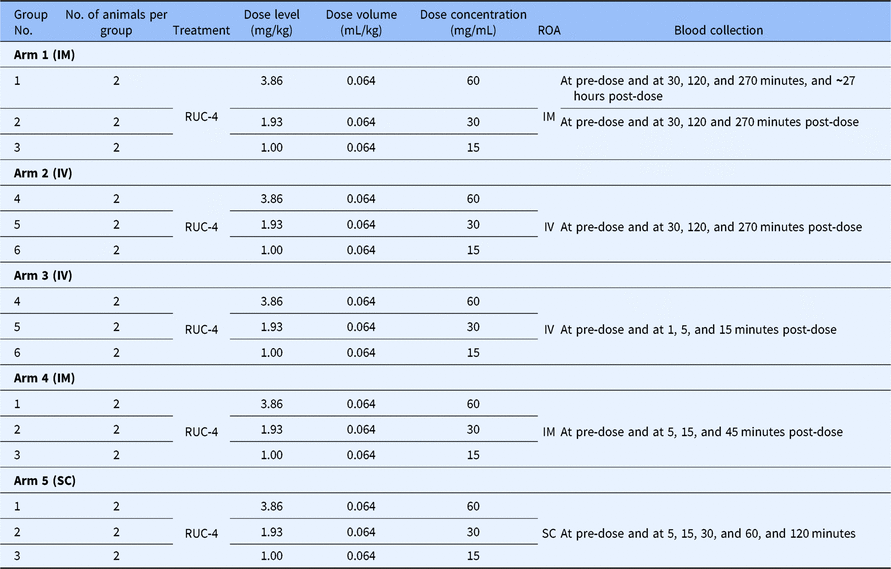
PK and PD data. The whole blood levels (µM), T½ values, bioavailability, and impact on platelet aggregation of RUC-4 in NHPs after IM and SC administration are given in Table 2 and Fig. 1. Both IM and SC RUC-4 reached dose-dependent peak levels within 5 minutes, with the exception of the 1.0 mg/kg SC dose, which peaked at 15 minutes. The C max after IM dosing was higher than after SC dosing, but all of the SC values between 30 and 120 minutes were higher than the IM values. The calculated T1/2 values were between 0.28 and 0.56 hours. The bioavailability of RUC-4 IM and SC compared to RUC-4 administered IV ranged between 0.60 and 0.88 at doses of 1.93 and 3.86 mg/kg, but there was greater variability in bioavailability at 1 mg/kg (1.2 IM and 0.55 SC). Platelet aggregation studies demonstrated >80% inhibition of the initial slope of aggregation in response to ADP in all samples in which the whole blood RUC-4 concentration was ≥ ~8 nM, with partial inhibition at lower concentrations.
Fig. 1. RUC-4 whole blood concentrations at various times after IM and SC injections, along with inhibition of ADP-induced platelet aggregation data after IM injection in NHPs.
Table 2. Whole blood levels (µM), half-lives, and bioavailability of RUC-4 in non-human primates after IM and SC administration
IM, intramuscular; SC, subcutaneous.
a Time points in which platelet aggregation studies were performed on animals treated with RUC-4 IM.
b Values of whole blood RUC-4 levels corresponding to an average of >80% inhibition of the initial slope of platelet aggregation in the two animals tested.
c Values of whole blood RUC-4 levels corresponding to an average of 0%–80% inhibition of the initial slope of platelet aggregation in the two animals tested.
Clinical pathology. The hematocrits and platelet counts of the animals before and after treatment with RUC-4 in arms 1, 2, and 5 are shown in Table 3. The reductions in hematocrit were considered consistent with the blood drawing from the animals. The platelet counts varied, but did not demonstrate obvious drug-related trends by dose or time after administration with the possible exception of a decrease in the count in response to the1.93 mg/kg IM RUC-4 dose in animal 4. Repeat RUC-4 doses of 1.93 mg/kg in this animal IV and SC in subsequent studies did not, however, elicit a similar response.
Table 3. Hematocrit and platelet count values before and at timed intervals after administering RUC-4 IM (A), IV (B), and SC (C)
IM, intramuscular; SC, subcutaneous; IV, intravenous
In Vitro Platelet Aggregation Studies in Healthy Human Volunteers
Effect of aspirin on the IC50 of RUC-4. The IC50 of untreated citrated PRP using 1.5 mM arachidonic acid was 61 ± 1 nM (Table 4). Aspirin eliminated arachidonic-acid-induced platelet aggregation, confirming its inhibition of thromboxane production.
Table 4. RUC-4 IC50 values (μM) for untreated platelets activated with arachidonic acid (1.5 mM) and untreated or aspirin-treated platelets activated with ADP (5 µM)

The IC50 of RUC-4 for ADP-induced aggregation of untreated platelets was 40 ± 9 nM, which was similar to the IC50 for aspirin-treated platelets (37 ± 5 nM; p = 0.39).
Effects of different anticoagulants on RUC-4 IC50 . The IC50s for RUC-4 in response to 20 µM TRAP activation were similar for PRP prepared from whole blood anticoagulated with heparin (15.8 U/ml) or PPACK (0.3 mM), 111 ± 7 and 122 ± 17 nM, respectively (p = 0.16; n = 4). In contrast, with PRP prepared from blood anticoagulated with sodium citrate (final concentration 0.38%), the IC50 was significantly lower, 66 ± 25 nM (p = 0.04 vs heparin and 0.05 vs PPACK; n = 4). Another series of experiments compared the RUC-4 IC50s in response to ADP activation (20 µM) of PRP prepared from blood anticoagulated with either sodium citrate (0.38%) or PPACK (0.1 mM). Once again, the IC50 was significantly higher in the PRP prepared from the PPACK-anticoagulated blood (102 ± 22 nM) compared to PRP from the citrate-anticoagulated blood (54 ± 13 nM; p <0.001: n = 5).
Experiments to assess the percentage of unoccupied αIIbβ3 receptors when platelets are treated with different doses of RUC-4. To assess the percentage of αIIbβ3 receptors that are not occupied by RUC-4 at different doses, we: (1) added FITC-labeled mAb PAC-1 (which selectively reacts with activated αIIbβ3 receptors) to PRP samples prepared from blood anticoagulated with PPACK (0.1 mM); (2) incubated the PRP with varying doses of RUC-4; (3) activated the receptor with ADP (20 µM); and (4) analyzed PAC-1 binding by flow cytometry. In parallel studies with the same PRP samples, we assessed the effect of the same concentrations of RUC-4 on platelet aggregation in response to ADP (20 µM) activation. The correlation between the inhibition of ADP-induced platelet aggregation and inhibition of PAC-1 binding for the five separate donors is shown in Fig. 2. With each donor, inhibition of platelet aggregation was discernible when there was ~40% or more inhibition of PAC-1 binding, presumably reflecting occupancy by RUC-4 of ~40% of the αIIbβ3 receptors. The inhibition of platelet aggregation was then an approximately linear function of inhibition of PAC-1 binding, reaching nearly complete inhibition of platelet aggregation when the inhibition of PAC-1 binding reached between 80% and 90%. The average correlation between inhibition of platelet aggregation and inhibition of PAC-1 binding was 0.96 (p < 0.0001).
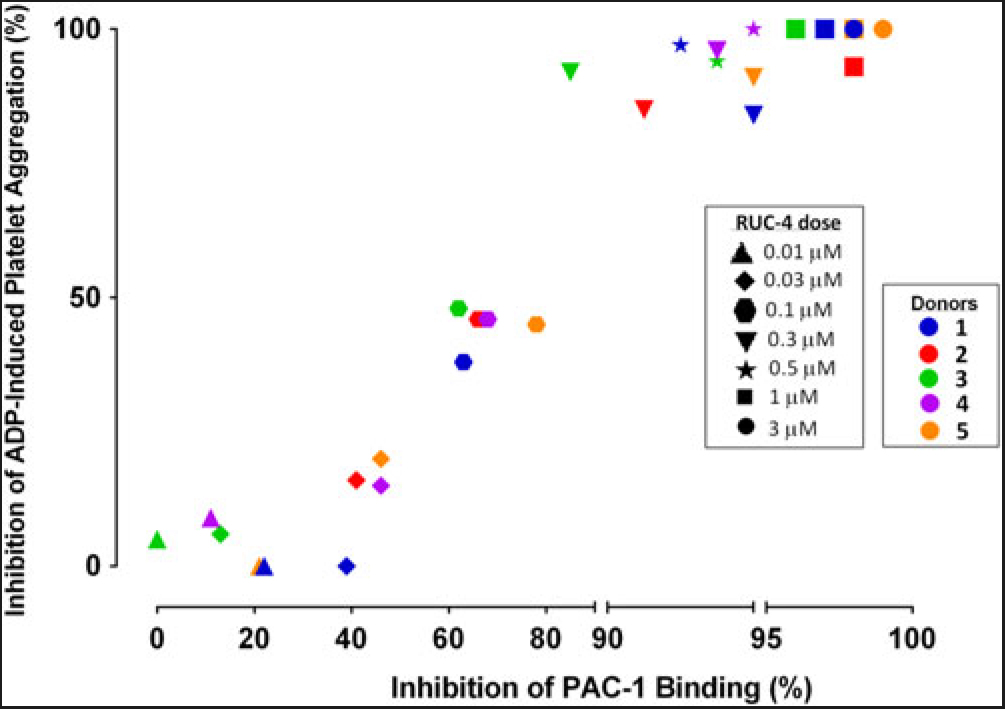
Fig. 2. Correlation between inhibition of platelet aggregation (%) and inhibition of PAC-1 binding (%) at different RUC-4 doses in samples from five volunteers.
Discussion
Early administration of αIIbβ3 antagonists, the most potent antiplatelet agents approved for human use, after STEMI symptom onset as a prelude to PCI improves both mortality and surrogate indicators of cardiac damage, including pre-PCI blood flow in the target vessel, time to ST segment resolution, left-ventricular ejection fraction, and distal embolization [Reference De Luca5–Reference Xu, Yin and Si7,Reference De Luca31]. The time to treatment is especially important as indicated by De Luca et al.’s finding that the cumulative mortality at approximately 11 years after STEMI in 814 patients was ~30% for those treated with an αIIbβ3 antagonist more than 178 minutes after symptom onset, ~16% for those treated between 101 and 178 minutes, ~10% in those treated between 65 and 110 minutes, and ~3% for those treated in less than 65 minutes (HR 1.41; 95% CI 1.02-2.21, p = 0.042 after correction for baseline confounding factors) [Reference De Luca6]. Further support for the importance of early therapy comes from a study of 10,730 patients with STEMI transported between 2015 and 2017 by emergency medical service personnel directly to a PCI-capable hospital in which 13% of the patients suffered a cardiac arrest or required intubation before the PCI was performed [Reference Jollis32]. It is challenging, however, to administer the currently available αIIbβ3 antagonists at the first point of medical contact because they all require IV administration as a bolus and a continuous infusion, the latter controlled by an electric pump. In addition, the current small molecule agents eptifibatide and tirofiban induce the receptor to undergo a major conformational change that may contribute to the development of thrombocytopenia in a small percentage of patients [Reference Aster15]. The conformational change also results in the receptor developing high affinity for its ligands, which may limit the efficacy of the agents as their blood concentrations drop and they elute from the receptors [Reference Cox18]. The oral P2Y12 antiplatelet agents, which are less potent than the αIIbβ3 antagonists, require hours to achieve their full antiplatelet effects, and studies in which they have been administered to STEMI patients as soon as possible have been disappointing when compared to the studies of the αIIbβ3 antagonists. [Reference Heestermans33,Reference Montalescot34] As a result, to achieve the antiplatelet benefits of the αIIbβ3 antagonists without the potentially negative effects of inducing the receptor to undergo the conformational change and without the need for IV administration by bolus and infusion, we developed RUC-4. We initially considered IM administration, but because SC administration is easier to deliver in the emergency setting, we now have assessed the feasibility of SC administration of RUC-4.
In accord with our previous studies with RUC-4-free base [Reference Li9], IM RUC-4 achieves peak blood levels in NHP within ~5 minutes, resulting in >80% inhibition of platelet aggregation that persists for several hours at a dose of 3.86 mg/kg. SC RUC-4 also achieves peak blood levels within ~5 minutes; while the peak concentrations are lower than those with IM RUC-4 at the same doses, the blood levels are more sustained than with IM administration. Thus, SC administration at 1 mg/kg produced for at least 2 hours whole blood levels exceeding those required for ≥80% inhibition of the initial slope of ADP-induced platelet aggregation after IM administration. These PK and PD data of SC RUC-4 are consistent with our goal of achieving rapid onset, short-term, high-potency inhibition of platelet aggregation as part of first point of care treatment of STEMI.
The inhibition of platelet aggregation correlated in general with the whole blood concentrations of RUC-4, but based on the NHP IC50 value, the extent of inhibition of aggregation was greater than expected at the lower levels of RUC-4 at the later time points. Potential explanations for this finding include incomplete measurement of RUC-4 by the bioanalytical assay, incomplete dissociation of RUC-4 from platelets by the lysing solution, or the generation of an RUC-4 metabolite that retains antiplatelet activity. Additional studies will be required to assess the contribution of each of these possibilities. In addition, since the IC50 value was obtained by adding RUC-4 to PRP, whereas the PK analysis is of whole blood, assuming a hematocrit of ~40%, the effective plasma concentration is 67% higher than the measured whole blood concentration.
Whereas aspirin is considered the standard of care for myocardial infarction, we plan to treat patients with STEMI with RUC-4 in combination with aspirin. To assess any pharmacologic drug–drug interactions between these agents, we assessed whether pretreating human platelets with aspirin would have an impact on RUC-4’s IC50 for inhibition of platelet aggregation. We found that treating platelets with aspirin at 0.83 mM, which was sufficient to eliminate platelet aggregation induced by arachidonic acid, did not materially affect the RUC-4 IC50. Future studies will need to address the potential for drug–drug PK and bleeding tendency and severity interactions between aspirin and RUC-4.
Previous studies demonstrated that the platelet inhibitory effects of αIIbβ3 antagonists differed when assessed with blood anticoagulated with citrate, which chelates divalent cations, including calcium and magnesium, and anticoagulants that do not chelate divalent cations, including heparin and PPACK [Reference Phillips25–Reference Kereiakes28]. In each case, the observed IC50 was lower with citrate-anticoagulated blood than with blood anticoagulated with non-chelating anticoagulants, although the agents varied considerably in the effect of citrate. As a result, we compared the RUC-4 IC50 in PRP prepared from blood anticoagulated with citrate, heparin, and PPACK. We also observed a lower IC50 in PRP prepared from blood anticoagulated with citrate, with the difference being approximately twofold. Previous investigators emphasized the importance of calcium chelation by citrate, but magnesium is also chelated by citrate [Reference Walser35] and magnesium occupies the MIDAS in β3 that is vital for ligand binding [Reference Zhu36]. Moreover, RUC-4, like RUC-2, displaces the magnesium ion from the MIDAS [Reference Li9,Reference Zhu13]. Thus, it is possible that the mechanism by which citrate sensitizes platelets to RUC-4 involved chelation of magnesium. Selecting the proper anticoagulant for studies of RUC-4’s antiplatelet effect in humans is very important since early studies of eptifibatide’s antiplatelet effects using citrate anticoagulant led to the selection of dosing regimens later found to be inadequate to sustain its antiplatelet effect when the studies were performed using PPACK as the anticoagulant [Reference Tcheng37].
We also studied the effect of RUC-4 on mAb PAC-1 binding as a surrogate indicator of receptor occupancy by RUC-4. The results were similar to those we previously obtained with the mAb 7E3, the parent mAb of abciximab, and mAb 10E5. [Reference Coller38–Reference Coller40] We were able to measure receptor occupancy directly with those antibodies by measuring the inhibition of the binding of radiolabeled antibody. The data are also similar to those observed with eptifibatide, where receptor occupancy was measured indirectly by the binding of an activation-dependent mAb [Reference Tcheng37]. Since we do not have radiolabeled RUC-4, we analyzed the ability of PAC-1 to bind to unoccupied receptors after activation by ADP. In particular, we found that inhibition of platelet aggregation begins when more than 40% of the receptors are blocked and that nearly complete inhibition of platelet aggregation requires ~80% or greater inhibition. The antithrombotic effect of mAb 7E3 was apparent at ~70% receptor blockade with less stringent thrombotic models and greater than ~80% inhibition with more stringent models [Reference Coller39–Reference Gold41].
We conclude that RUC-4’s PK and PD in NHPs after SC administration support its potential as a potent, rapidly-acting, short-term antiplatelet therapy for first-point-of-medical-contact therapy of STEMI. In addition, aspirin does not affect RUC-4’s IC50, so combined use of the two agents for STEMI does not have to take into consideration a functional drug–drug interaction. Inhibition of platelet aggregation by RUC-4 is mildly enhanced by citrate anticoagulation and so monitoring of RUC-4’s antiplatelet effect should be conducted with blood anticoagulated with PPACK or another non-chelating anticoagulant. Finally, the relationship between the percentage of receptors blocked by RUC-4 and inhibition of platelet aggregation is very similar to that observed with other antiplatelet agents that target αIIbβ3. Based on these results, RUC-4 has now progressed to evaluation in separate toxicology and PK studies in mice and similar studies in NHPs in the presence and absence of aspirin [Reference Grimes42,Reference Hebert43].
Acknowledgments
The research was supported in part by grant 19278 from the National Heart, Lung and Blood Institute and grant # UL1 TR001866 from the National Center for Advancing Translational Sciences (NCATS, National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program, National Institutes of Health; The Robertson Therapeutic Development Fund; and funds from Stony Brook University. We express our appreciation to the nursing staff of the Rockefeller University Hospital for their assistance in obtaining blood samples from the human donors.
Disclosures
In accord with Federal law and the policies of the Research Foundation of the State University of New York, Mount Sinai School of Medicine, and Rockefeller University, respectively, B. S. Coller has royalty interests in abciximab (Centocor), the VerifyNow assays (Accumetrics), and the RUC compounds. C. J. Thomas also has a royalty interest in the RUC compounds. In accord with Rockefeller University’s policies, B. S. Coller is a consultant to, and has an equity interest in, CeleCor Therapeutics, which has licensed the RUC compounds from Rockefeller University.






 Open access
Open access