


It has been estimated that, worldwide, nearly three million children under the age of 5 years have clinical symptoms (night blindness and xerophthalmia) of vitamin A deficiency (VAD), and more than 200 million people are considered vitamin A deficient(1). In more than half of all countries, VAD is a significant public health issue, particularly in Africa and parts of Asia, where, according to the WHO, it causes blindness in more than 250 000 children every year. Subclinical VAD compromises immune function(Reference Sommer and West2) and is a major contributor to mortality in affected adults and children.
Strategies to combat VAD have broadly followed three main themes(Reference Sommer and West2, Reference Gillespie and Mason3): supplementation; fortification; dietary improvement. The latter approach is probably the most sustainable locally and, therefore, the best long-term method of controlling the prevalence of VAD, and eventually eliminating it. However, it will fail if local agriculture cannot produce foods with adequate vitamin A or precursor content, or if vitamin A in the ingested foodstuff is not optimally absorbed and utilised by the body.
Vitamin A and its precursor carotenoids are fat-soluble; sources from foods of animal origin are in the form of retinyl esters (principally palmitate), while vegetables provide provitamin A principally in the form of β-carotene, although α-carotene, γ-carotene and β-cryptoxanthin also make a contribution. These compounds exit the stomach and enter the small intestine in bulk liquid droplets. The retinyl esters are enzymatically hydrolysed to retinol, and then absorbed by passive diffusion into the enterocytes along with the carotenoid sources of vitamin A. In the enterocytes, retinol is re-esterified to palmitate, incorporated into chylomicrons and secreted into the lymphatic system. The carotenoids, on the other hand, may be directly incorporated into chylomicrons, or they may be cleaved to form retinyl esters before incorporation. Some of the carotenoids remain in the enterocytes and are lost as the cells are removed. Because of this complex metabolism, less retinol is derived from the carotenoids than might be expected from stoichiometric considerations of the molecular structure. The ratio of the amount of retinol obtained for normal physiological function to the quantity of β-carotene ingested is termed bioavailability(Reference Jackson4). In the case of vitamin A derived from the carotenoids, the bioavailability clearly incorporates a term describing the bioconversion (the definitions of bioavailability and bioconversion are not consistent within the literature. Some authors used bioconversion as a blanket term to cover both availability and subsequent conversion(Reference Castenmiller and West5, Reference van Lieshout, West and van Breemen6)). The bioavailability of vitamin A from a food depends on a number of factors including the species of carotenoid from which it is derived, the chemical form (e.g. free or as esters) in which that carotenoid is present, the amount of carotenoid present in the food, the matrix in which it exists, the presence of other food constituents which enhance or inhibit absorption, and the nutritional status health, and genetic profile of the subject. In general, it is not expected that these factors will operate independently, and considerable interactions are likely to determine the bioavailability of vitamin A from a particular carotenoid in a given subject.
Given all of these factors, probably the simplest manipulation to improve vitamin A bioavailability from carotenoids is meal preparation. The choice of vegetable may be restricted by the local agricultural conditions, but its use as a source of vitamin A can be optimised by considering the way in which it is prepared for the table. It has been noted that both provitamin A carotenoids and vitamin A itself are fat-soluble, and therefore it is reasonable to predict that carotenoids are more likely to be utilised if they are presented in an environment with sufficient fat to assist in their uptake. On this basis, it has been predicted and demonstrated that fried vegetables are better sources of vitamin A than raw ones, whereby the adoption of a diet where a higher fraction of the vegetables is fried might be beneficial in populations at risk of VAD. The object of the present study was to compare the bioavailability of vitamin A from raw and stir-fried carrots using stable isotope tracers. In order to ensure that true bioavailability from the vegetable matrix was determined, it was decided that the carrots should be intrinsically labelled, and this was simply achieved by irrigating them during growth with 2H-enriched water. The carrots were then prepared for eating, either raw or stir-fried, and the kinetics of the labelled carotenoids and retinol established in the plasma of healthy volunteers. To control for inter- and intra-individual variation, we gave oral doses of synthetic extrinsic 13C-labelled β-carotene and [2H4]retinol simultaneously with the carrot meal.
Materials and methods
Reagents and equipment
Reagents and apparatus were obtained from the following suppliers:
Aldrich Chemical Company Inc., Milwaukee, WI, USA – 2H2O (99 %).
ARC Laboratories BV, Apeldoorn, The Netherlands – 8,8′,9,9′,10,10′,11,11′,12,12′,13,13′,14,14′,15,15′,19,19′,20,20′-[13C20]β-carotene, (all-E)-([13C20]β-carotene); 10,19,19,19-[2H4]retinol acetate ([2H4]retinol acetate).
CaroteNature GmbH, Lupsingen, Switzerland – α-carotene, all-trans; β-cryptoxanthin.
Fisher Scientific UK Limited, Leicestershire, UK – acetonitrile (ACN, HPLC grade); magnesium carbonate; MeOH (HPLC grade).
IKA Werke GmbH, Staufen, Germany – homogeniser, Ultra Turrax model T25.
Riedel-de Haën, Seelze, Germany – absolute ethanol.
Sigma Aldrich Limited, Dorset, UK – butylated hydroxytoluene; (all-E)-β-carotene, synthetic; ethyl-β-apo-8′-carotenoate; petroleum diethyl ether (35–60°C fraction) spectrophotometric grade; platinum (IV) oxide; retinol acetate, all-trans, synthetic.
VWR International Limited, Dorset, UK – dichloromethane (HPLC grade); n-heptane (HPLC grade); hexane (HPLC grade); tetrahydrofuran.
Intrinsic labelling of carrots
2H2O (99 %) was diluted with local tap water to give a 2 % solution. Carrot seeds (F1 Healthmaster Daucus carota, Autumn King) were sown in Levington potting compost and grown in an unheated greenhouse. Not < 1 week after germination, the hole in the bottom of the pot was taped over, and the top covered with a plastic bag, which had been pierced with a small hole. These measures were adopted to minimise the rate of water evaporation while allowing some transpiration to occur. The plants were watered daily with 2H2O for 7 weeks, and then the carrots were harvested.
The carrots were washed and the ends were cut, before being peeled and then blanched for 6 min(Reference Kendall7, Reference Desobry, Netto and Labuza8), after which they were plunged into ice water to halt the cooking process. The carrots were then homogenised in a food processor, divided into portions of 110 g, which were individually wrapped in Al foil and placed in plastic bags for storage at − 20°C until required.
Carotenoid analysis of carrots
The carotenoid content of the grown contents was determined by HPLC of a lipid extract (Fig. 1), where the sample processing procedure is divided into segments A–I, as follows:
(a) The sample extraction was achieved using the method described by Hart & Scott(Reference Hart and Scott9). Duplicate samples (10 g) of carrot were homogenised together with 1 g MgCO3 and 50 ml (1:1, v/v) tetrahydrofuran–MeOH containing 165 μg (all-E)-ethyl-β-apo-8′-carotenoate as an internal standard.
(b) The dried extract was dissolved in 20 ml dichloromethane, and a 200 ml aliquot diluted in 10 ml mobile phase, ACN–MeOH–dichloromethane (44:44:12 %, v/v/v). Isocratic separation was achieved using a Waters Alliance 2695 Separation Module equipped with a 4·6 mm × 100 mm reverse-phase C18 cartridge column with a 3·5 μm particle size and a 10 nm pore size (Waters UK Ltd, Elstree, Herts, UK). The column was thermostatically controlled at 23·0 ± 0·1°C for the stability of retention time. Detection was accomplished by a Waters 996 photodiode array (Waters Limited) operating in the range of 280–480 nm. Quantification was achieved by calibration with a standard solution containing α-carotene, β-carotene and β-cryptoxanthin at concentrations of the order of 0·4 μmol/l, with retinol acetate and ethyl-β-apo-8′-carotenoate as internal standards.
(c) For the isotopic investigation, the β-carotene fraction was collected and dried under N2 at room temperature. The perhydro derivative was then prepared based on published methods(Reference Yao, Liang and Trahanovsky10, Reference Parker, Brenna and Swanson11), by dissolving the residue in 0·5 ml hexane and transferring to sample vials containing a platinum (IV) oxide catalyst. The vials were flushed through and filled with hydrogen at 2 bar, and then left in a heating block at 65°C in the dark for 16 h. The tubes were washed three times with 200 μl hexane, which was then transferred to amber silanised GC vials, dried under N2 at room temperature, and redissolved in 10 μl hexane before analysis.

Fig. 1 Schematic diagram illustrating the key steps taken to extract carotenoids from carrots and carotenoids and retinol from blood plasma, for their eventual quantitative measurement on HPLC and qualitative measurement on GC–MS.
GC/MS was performed using a 6890 GC coupled to a 5973 mass-selective detector (Agilent Technologies, West Lothian, UK) with an on-column injector. The injector was fitted with an Agilent 1·0 m × 0·530 mm deactivated fused silica retention gap. The retention gap was connected via a metal union to a 15 m × 0·25 mm (inner diameter) fused silica capillary column coated with a DB1-MS stationary phase (0·25 μm in thickness; J&W Scientific, Agilent Technologies UK Ltd, Stockport, Cheshire, UK). Chromatographic separation was achieved with a four-stage temperature programme: 60–265°C at 25°C/min, followed by 265–300°C at 10°C/min, before a 4 min isothermal step. Finally, the column was ramped to 325°C over a final minute to complete elution of remaining materials. The isotopomer ratios were measured in selected ion monitoring mode of the mass cluster 558–562 m/z using the peak-fitting method of Bluck & Coward(Reference Bluck and Coward12).
Hydrogenation transforms the carotene, C40H56 (molecular weight 536), to the perhydro compound C40H78 (molecular weight 558), adding twenty-two hydrogen atoms (and in the process destroying the chiral centre that distinguishes α- from β-carotene). The cracking pattern of the molecular cluster can be analysed by assuming that the molecule is made up of two species of hydrogen, one comprising fifty-six sites with fractional abundance F a, and the other twenty-two sites at abundance F b, in addition to the carbon with abundance F C. Elementary permutation and combination theory allows the isotopomer distribution to be calculated(Reference Bluck and Volmer13) to obtain expressions for the mass ratios 559/558 (M+1), 560/598 (M+2) and 561/598 (M+3) in terms of the elemental fractional abundances (see Appendix 1). It is then assumed that the carbon and the b-hydrogens are at natural abundance (F C = 0·0111, F b = 0·00 015)(Reference Bohlke, de Laeter and De Bievre14), and a value for F a obtained by least squares fitting the ratios to the experimentally observed values.
In vivo determination of bioavailability of carotenoids from carrots
Subjects
A total of five healthy subjects were recruited to the study by advertisement. The study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects were approved by the Cambridge Local Research Ethics Committee (02/069 approved on 2 April 2002) and informed written consent was obtained from all subjects. Potential recruits were screened at an interview using a medical questionnaire, and excluded if they were smokers or heavy users of alcohol, taking nutrient supplements containing vitamin A, suffering from significant renal, liver or respiratory disease or had any disorder, which might interfere with gastrointestinal function, or had been diagnosed with diabetes. Women who were pregnant or intending to become pregnant during the course of the study were also excluded, as were those who were breast-feeding.
Overall protocol
The study was intended as a proof of principle to determine a methodology for investigating the effects of cooking method and overall meal composition on the bioavailability of vitamin A from a particular meal component. For the purposes of the study, a comparison of the bioavailability of raw and stir-fried carrots was made, following the hypothesis that the lipid-soluble carotenoids would be more bioavailable from a meal matrix containing added fat in the form of a cooking vegetable oil. The subjects were therefore asked to participate in two sessions of testing, separated by a period of 4 weeks. On the first occasion, they were randomly assigned to consume a test meal of either raw or stir-fried carrots, with the alternative meal being given on the second visit.
Meal study protocol
The subjects were asked to attend the volunteer suite at Human Nutrition Research (HNR) on the morning of the 1st day of the meal study having fasted for 12 h. Their height and weight were recorded to the nearest 0·1 kg and 0·01 m, and 10 ml of basal blood samples were collected. Blood samples in the present study were taken by venepuncture into 9 ml and 4·9 ml S-Monovette lithium heparin tubes (Sarstedt Ltd, Beaumont Leys, Leics, UK). The subject then consumed a single bolus dose of 100 g of the intrinsically labelled carrots (either raw or stir-fried in 10·5 ml groundnut oil), with 3·6 μmol of [13C20]β-carotene in 2·1 ml groundnut oil, and 4·8 μmol [2H4]retinol acetate in 0·75 ml groundnut oil. The intrinsically labelled carrots were minimally processed by blanching and homogenisation before storage at − 20°C. The carrot meals that had no further processing carried out to the carrots will be described as ‘raw’ in the present study, while those carrots that had been stir-fried, will be described as ‘stir-fried’.
Further 10 ml of blood samples were taken at 6, 8, 10 and 13 h after the meal was given. The subjects then left the HNR unit but returned on each of the following 3 d for 10 ml of blood samples to be taken at 24, 28, 32, 36, 48, 54 and 72 h post-dosing. Subjects were allowed to eat and drink normally from 4 h after consuming the carrots, but they were asked to avoid foods with high carotenoid or vitamin A content (carrots, carrot juices, mangoes, spinach, oily fish and foods cooked in red palm oil) for the 3 d blood sampling period. Written guidance for this was provided.
Measurement of carotenoid and retinoid concentrations in plasma
Plasma was separated from the blood by centrifugation, and stored at − 80°C until required for analysis. Quantitative determination of (all-E)-α-carotene, (all-E)-β-carotene, β-cryptoxanthin, retinol palmitate and total retinol was achieved following the method of Thurnham et al. (Reference Thurnham, Smith and Flora15), except that the two internal standards, 0·50 nmol trans-ethyl-β-apo-8′-carotenoate and 1·2 nmol all-trans-retinol acetate, were used in place of tocopherol acetate. The final dry extract was reconstituted first in 30 μl dichloromethane and then 220 μl ACN–MeOH (1:1) were added.
Quantitative HPLC was performed using isocratic separation on a Waters Alliance 2695 Separation Module equipped with a thermostatically controlled 4·6 mm × 100 mm reverse-phase C18 cartridge column with a 3·5 μm particle size and a 10 nm pore size (Waters Limited). Dual-channel photodiode array detection (Waters Limited) at 450 nm for carotenoids and at 325 nm for retinoids was used, and quantification was achieved by comparison with the National Institute of Standards and Technology Standard Reference Material 968c(Reference Thomas, Kline and Gill16).
Preparation of carotenoids and retinol for isotopic analysis
The overall method is shown schematically in Fig. 1. With reference to this figure, the following six steps are described in detail as follows:
(d) A 2·5 ml sample of plasma was split into three aliquots, and each was deproteinised with ethanol and the lipids were extracted with hexane. The extracts were combined and dried at room temperature under N2 gas.
(e) The dry residue was then redissolved in 600 μl of 76:24 % (v/v) MeOH–dichloromethane. Semi-preparative chromatography was performed on 200 μl aliquots using a Waters 515 pump (Waters UK Ltd) and a Vydac TP201 reverse-phase column (W. R. Grace & Co., Deerfield, IL, USA). A Waters 2487 UV–visible detector (Waters UK Ltd) was used to identify fractions absorbing at 325 and 450 nm for fraction collection. Unesterified retinol was collected separately from the carotenoids and retinyl esters.
(f) After drying under N2, the carotenoid and retinyl ester fraction was redissolved in 2 ml of 2 % ethanolic potassium hydroxide. Saponification was achieved in 25 min at 45°C after which the samples were cooled and 0·5 ml of deionised water were added. The carotenoids and retinol were extracted with three washes of hexane (1 ml), which were then combined and dried completely.
(g) The samples were reconstituted in 120 μl of 90:10 % (v/v) MeOH–dichloromethane and separated isocratically on a Vydac 201TP54 column (W. R. Grace & Co.) using 90:10 % (v/v) MeOH–dichloromethane as the mobile phase. The carotene fraction was then hydrogenated ready for GC–MS analysis as described for the carrot extracts.
(h) The retinol fraction was dried, and then redissolved in 120 μl absolute ethanol before further purification using a Waters C18-symmetry cartridge column with gradient elution as follows: ACN–tetrahydrofuran–1 % aqueous ammonium acetate (50:20:30, v/v/v, in water) was used for 6 min, followed by an 8 min linear gradient introducing 50 % ACN–tetrahydrofuran–1 % aqueous ammonium chloride (40:44:6, v/v/v). These conditions were held for 5 min before the second solvent was linearly increased to 100 %. These conditions were held for 8 min, and then a 1 min linear transition to the original solvent was made. The retinol fraction was collected and 2 ml chloroform added. After centrifugation, the aqueous layer was removed and the organic solvents were evaporated under N2.
(i) Retinol was washed with 2 ml ethanol to absorb any remaining water, and then dried before trimethylsilyl diethyl ether was formed using BSTFA (N,O-bis (trimethylsilyl) trifluoroacetamide) in pyridine(Reference Knapp17). Trimethylsilyl diethyl ether was dried and reconstituted in 100 μl of derivatising agent ready for GC–MS analysis.
GC–MS was performed using a 6890 GC series system (Agilent Technologies), with an on-column injection of 1 μl of the sample. Chromatographic separation was achieved on a DB1-MS column (J&W Scientific), with a four-stage temperature programme: 60–200°C at 25°C/min, followed by 200–235°C at 6°C/min, then increasing to 330°C at 30°C/min and held at 340°C for 2·4 min to elute any remaining materials. An ion source temperature of 150°C and a quadrupole temperature of 120°C were used. The mass spectrometer was used in low-resolution mode, with signal in the range of ± 0·2 m/z being integrated for each mass measured. Tuning, including a mass scale calibration, was performed before each batch of samples were analysed. The isotopomer ratios were measured in selected ion monitoring mode of the mass cluster 359–371 m/z using the peak-fitting method of Bluck & Coward(Reference Bluck and Coward12).
It was determined that the CV on repeated M+1/M measurements for either hydrogenated β-carotene or derivatised retinol on the same plasma sample (internal error) was < 0·5 %. The CV on M+1/M measurements of different extractions was 1–1·5 %.
Isotopic analysis of perhydro β-carotene
There are expected to be three distinct isotopic species of β-carotene in the plasma samples: the pre-existing naturally abundant species that labelled randomly with 2H obtained from the carrots, and [13C20]β-carotene given as a reference. After hydrogenation, the measurable peaks in the molecular ion cluster of the first two species are in the range 558–561 m/z, while for the latter, the corresponding signal is at 578–580 m/z. Using the notation described in Table 1, the mole fractions of all three species can be deduced from mass spectral intensities from
where the matrices
![\begin{eqnarray} { C _{ = } } = \left [{\begin{array}{ccc}( A _{U} - A _{Y}) R _{559} - ( B _{U} - B _{Y}) & A _{U} R _{559} - B _{U} \\ ( B _{U} - B _{Y}) R _{560} - ( C _{U} - C _{Y}) & A _{U} R _{560} - C _{U} \\ ( C _{U} - C _{Y}) R _{561} - ( D _{U} - D _{Y}) & A _{U} R _{561} - D _{U} \\ ( A _{U} - A _{D}) R _{578} & A _{U} R _{578} + A _{C} \\ ( A _{U} - A _{D}) R _{579} & A _{U} R _{579} + B _{C} \\ ( A _{U} - A _{D}) R _{580} & A _{U} R _{580} + C _{C} \\ \end{array} }\right ], \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU2.gif?pub-status=live)
![\begin{eqnarray} { H _{\lowbar } } = \left [{\begin{array}{ccc} A _{U} R _{559} - B _{U} \\ A _{U} R _{560} - C _{U} \\ A _{U} R _{561} - D _{U} \\ A _{U} R _{578} \\ A _{U} R _{579} \\ A _{U} R _{580} \\ \end{array} }\right ] \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU3.gif?pub-status=live)
and
Table 1 Contributions of each of the three isotopically distinct β-carotene species in plasma to the overall mass spectrum: the pre-existing naturally abundant species (unlabelled) that labelled randomly with 2H obtained from the carrots (from carrots), and [13C20]β-carotene given as a reference (13C-labelled)
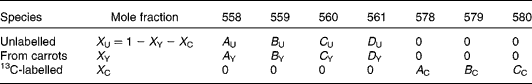
(see Appendix 2).
The coefficients adopted for the carrot β-carotene (A Y, B Y, C Y, D Y) were those obtained in the calculation of the degree of intrinsic labelling (see the Results section). Those for the unlabelled (basal) and [13C20]β-carotene were calculated in the same way, but assuming that the element with the major deviation from fractional abundance in these compounds was carbon.
Isotopic analysis of trimethylsilyl-retinol
A similar approach was adopted for trimethylsilyl-retinol, except now there are four isotopically distinct sources to consider: the pre-existing (basal) unlabelled retinol; that due to cleavage of the carrot β-carotene; some due to cleavage of [13C20]β-carotene ([13C10]retinol); [2H4]retinol used as a standard. There are twelve mass numbers that are useful in the spectrum of trimethylsilyl-retinol in the range 358–371 m/z. The significant contributions of each species are indicated in Table 2. The least-squares solution to the eleven equations in three unknown mole fractions is the solution to:
![\begin{eqnarray} \left [{\begin{array}{ccc}[ B _{U} - B _{Y} - ( A _{U} - A _{Y}) R _{359}] & ( B _{U} - A _{U} R _{359}) & ( B _{U} - A _{U} R _{359}) \\ [ C _{U} - C _{Y} - ( A _{U} - A _{Y}) R _{360}] & ( C _{U} - A _{U} R _{360}) & ( C _{U} - A _{U} R _{360}) \\ [ D _{U} - D _{Y} - ( A _{U} - A _{Y}) R _{361}] & ( D _{U} - A _{U} R _{361}) & ( D _{U} - A _{U} R _{361}) \\ ( A _{U} - A _{Y}) R _{362} & A _{U} R _{362} & ( A _{D} + A _{U} R _{362}) \\ ( A _{U} - A _{Y}) R _{363} & A _{U} R _{363} & ( B _{D} + A _{U} R _{363}) \\ ( A _{U} - A _{Y}) R _{364} & A _{U} R _{364} & ( C _{D} + A _{U} R _{364}) \\ ( A _{U} - A _{Y}) R _{365} & A _{U} R _{365} & ( D _{D} + A _{U} R _{365}) \\ ( A _{U} - A _{Y}) R _{368} & ( A _{C} + A _{U} R _{368}) & A _{U} R _{368} \\ ( A _{U} - A _{Y}) R _{369} & ( B _{C} + A _{U} R _{369}) & A _{U} R _{369} \\ ( A _{U} - A _{Y}) R _{370} & ( C _{C} + A _{U} R _{370}) & A _{U} R _{370} \\ ( A _{U} - A _{Y}) R _{371} & ( D _{C} + A _{U} R _{371}) & A _{U} R _{371} \\ \end{array} }\right ]\left [{\begin{array}{ccc} X _{Y} \\ X _{C} \\ X _{D} \\ \end{array} }\right ] = \left [{\begin{array}{ccc} B _{U} - A _{U} R _{359} \\ C _{U} - A _{U} R _{360} \\ D _{U} - A _{U} R _{361} \\ A _{U} R _{362} \\ A _{U} R _{573} \\ A _{U} R _{364} \\ A _{U} R _{364} \\ A _{U} R _{365} \\ A _{U} R _{369} \\ A _{U} R _{370} \\ A _{U} R _{371} \\ \end{array} }\right ]. \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary-alt:20160626131725-85430-mediumThumb-S000711451100451X_eqnU5.jpg?pub-status=live)
Table 2 Contributions of each of the four isotopically distinct retinol species in plasma to the overall mass spectrum: the pre-existing (basal) unlabelled retinol (unlabelled); that due to cleavage of the carrot β-carotene (from carrot β-carotene); some due to cleavage of [13C20]β-carotene, namely [13C10]retinol (from [13C20]β-carotene) and [2H4]retinol used as a standard (from [2H4]retinol acetate)

In this instance, it is not useful to adopt the same procedure to calculate the coefficients of the mass spectrum as the addition of Si to the molecule for analysis with large isotopic contributions of its own gives additional complexity, which cannot be resolved. On this basis, the mass spectral coefficients A, B, etc. for the labelled species are calculated from the atomic abundances(Reference Bluck and Volmer13), and are given in Table 2. For the unlabelled (pre-existing) retinal, the coefficient A U is calculated in the same way, and B U, C U and D U derived from it using the measured basal isotopomer ratios.
Kinetic modelling
Simple compartmental models were used to interpret the observed kinetics of retinyl palmitate, retinol and β-carotene. In each of the three cases, the simplest model that accounted for the observed behaviour was employed. Parameter determination was accomplished by minimising the sum of the squared residuals between fitted and experimental data using the non-linear optimisation function in Excel (Microsoft Inc., Redmond, WA, USA).
Retinyl palmitate
Mean tracer retinyl palmitate kinetics in the period 0·25–1 d after dosing were described by:
where m RE is the total quantity of retinyl palmitate delivered to the sampled pool and k RE is the fractional rate constant of disappearance. This equation describes the first-order loss of a bolus quantity from a single pool (Fig. 2(a)). This pool was identified as the plasma, and therefore its volume of distribution, V R, was estimated by first calculating body surface area from weight and height(Reference Haycock, Schwartz and Wisotsky18) and then using the prediction equations proposed by Hurley(Reference Hurley19). The combined expressions are:
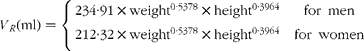
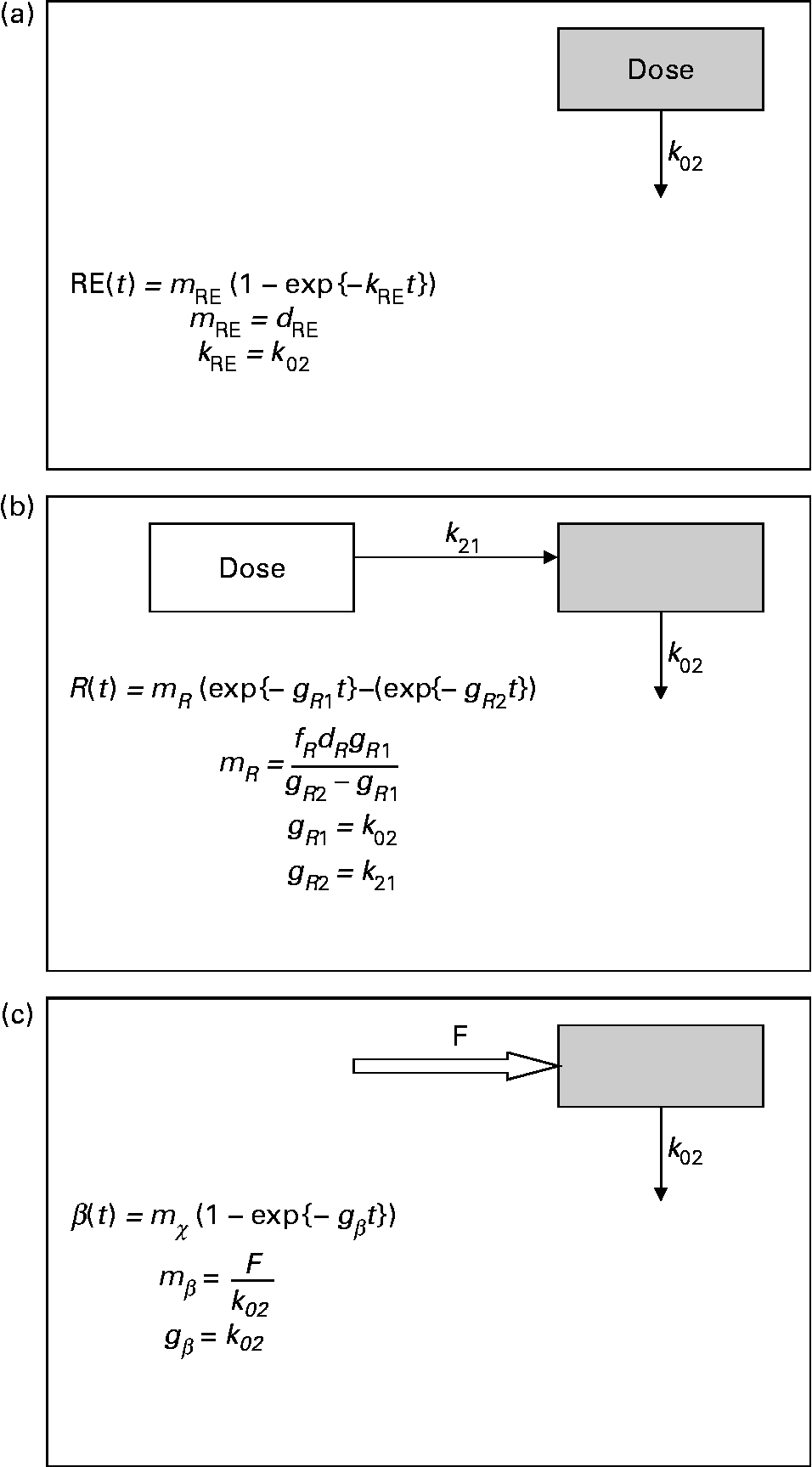
Fig. 2 Parsimonious models used: (a) model of retinyl ester kinetics, (b) model of retinol tracer kinetics and (c) model of β-carotene tracer kinetics. In each case, ![]() indicates the sampled compartment. The equations relate the fundamental fractional rate constants in the model to the experimentally determined parameters. retinol equivalent
indicates the sampled compartment. The equations relate the fundamental fractional rate constants in the model to the experimentally determined parameters. retinol equivalent
Retinol
For tracer retinal, a dual exponential function was needed to describe the observed kinetics:
where m R, g R1, g R2 and τ are constants, the latter representing a delay between dosing and the appearance of the dose in the plasma. This expression is characteristic of a pool fed by a pool to which the dose has been delivered, and with a route of irreversible loss (Fig. 2(b)).
β-Carotene
The time courses of plasma tracer β-carotene, obtained by multiplying the total plasma concentration from HPLC by the mole fraction obtained from GC–MS, are described by the single exponential function:
where m β and k β are constants. This expression is typical of a compartment with a constant flux input, and first-order irreversible loss (Fig. 2(c)). When performing the optimisation for this model, the data obtained in the first 12 h after dosing were not included, since this is dominated by the transient chylomicron transport of newly absorbed β-carotene to the liver.
Results
Carotenoid content of the intrinsically labelled carrots
Analysis of the raw carrots, after minimal processing of blanching and homogenisation, indicated that each 100 g (wet weight) contained 3·45 (sd 0·44) mg (6·4 (sd 0·5) μmol) α-carotene and 3·79 (sd 0·44) mg (7·1 (sd 0·8) μmol) β-carotene, approximately the same levels (3 mg/100 g and 6 mg/100 g, respectively) are reported elsewhere for young carrots(Reference Agency20). The fractional abundance of 2H in β-carotene obtained by combining five individual estimates was 0·636 (sd 0·036) atom%, representing an enrichment of 0·621 % above natural abundance.
This labelling pattern produces a theoretical mass spectrum with relative abundances of 0·4461 (sd 0·0041) at 558 m/z, 0·3616 (sd 0·0008) at 559 m/z, 0·1450 (sd 0·0020) at 560 m/z and 0·0384 (sd 0·0010) at 561 m/z (isotopomer ratios R 559 = 81·1 (sd 0·9) %, R 560 = 32·5 (sd 0·7) % and R 561 = 8·6 (sd 0·3) %), which was used in the speciation of plasma β-carotene.
Volunteer characteristics
Volunteer characteristics are given in Table 3. Of the subjects, one (D) did not complete one of the days' protocol and was therefore excluded from the analysis. Plasma volume from weight and height considerations was estimated to be 2·9 (sd 0·2) litres. The meals were given in randomised order. No difference in vitamin A status, judged by pre-meal plasma concentrations of β-carotene or retinol, was noted.
Table 3 Volunteers' age, anthropometric measurements, and study plasma basal carotenoid and retinol concentration values in each 4 d study period

Plasma carotenoid and retinoid concentrations after meal ingestion
There was no detectable increase in plasma α- or β-carotene concentration due to either meal protocol. Although plasma retinol was similarly unperturbed, retinyl palmitate showed a significant increase after both meals. Generally speaking, the peak value of retinyl palmitate had already occurred before 6 h after dosing, when the increments were 826 (sd 145) nmol/l (P < 0·01) and 888 (sd 243) nmol/l (P~0·07) for the raw and stir-fried meals, respectively.
The averaged values obtained from the four subjects were fitted to an exponential (equation 1) in the period 0·25–1 d, giving k RE equal to 2·8 (sd 0·4) per d after the raw carrot meal and 3·2 (sd 0·7) per d after the stir-fry. When extrapolated back to zero time, the intercepts of the curves were 1·3 and 1·1 μmol/l, which when combined with the estimates of plasma volume correspond to 3·0 and 3·2 μmol for the raw meal and the stir-fried meal, respectively.
Plasma retinol kinetics
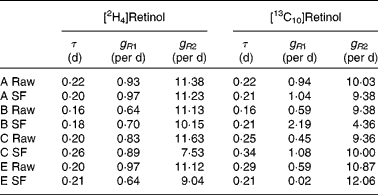
Typical retinol kinetics obtained from the stable isotope data are shown in Fig. 3. The values obtained for the macroparameters τ, g R1 and g R2 are given in Table 4. Since there are no significant differences in these three parameters between the retinol species, each test was remodelled assuming common values as this gives additional strength to the analysis. Using this approach, the self-consistent parameters for the raw carrot meal were found to be τ = 0·19 (sd 0·01) d, g R1 = 0·77 (sd 0·07) per d, g R2 = 10·8 (sd 0·3) per d, m R(2H4) = 0·193 (sd 0·012) μmol/l and m R(13C10) = 0·065 (sd 0·017) μmol/l, while for the stir-fried meal, the values τ = 0·22 (sd 0·02) d, g R1 = 0·81 (sd 0·09) per d, g R2 = 10·3 (sd 1·1) per d, m R(2H4) = 0·206 (sd 0·020) μmol/l and m R(13C10) = 0·055 (sd 0·017) μmol/l were found.

Fig. 3 Typical plasma retinol concentration v. time profiles for the five subjects over a 4 d study period, after consumption of a single bolus meal of 100 g of the intrinsically labelled carrots (either raw or stir-fried in 10·5 ml groundnut oil), with 4·8 μmol [2H4]retinol acetate and 3·6 μmol [13C20]β-carotene in groundnut oil. Plasma [2H4]retinol (■) and [13C10]retinol (![]() ) are derived from the [2H4]retinol acetate and [13C20]β-carotene doses, respectively. ○, Carrot retinol.
) are derived from the [2H4]retinol acetate and [13C20]β-carotene doses, respectively. ○, Carrot retinol.
Table 4 Retinol kinetics obtained for each subject (A–E) after consumption of either the raw or stir-fried (SF) meals
Plasma carotenoid kinetics
A typical time course of plasma β-carotene is shown in Fig. 4. The early stages (t < 0·5 d) represent β-carotene being carried to the liver in the chylomicron fraction of the lipid. The lifetime of chylomicrons is relatively short, and the profile of the curve in this early phase is governed both by the length of the period of β-carotene absorption (which in turn is a function of gastric-emptying rate and rate of transport through the enterocytes) and the rate of the hepatic breakdown of chylomicrons. The shape of the curve after the initial transient event approximates to an exponential rise to a steady plateau, typical of the kinetics of steady release into a single compartment with a small fractional rate of loss. Adopting this model and fitting using equation 4 in terms of m β and k β, a similar observation was made to that for retinol inasmuch as the fractional rate constant was independent of isotopic species, and therefore final analysis was performed using a common value of k β for the two species. Its value was found to be 4·3 (sd 1·7) per d for the raw carrot meal and 4·1 (sd 0·4) per d for the stir-fried meal. For [13C20]β-carotene, m β was found to be 0·051 (sd 0·007) μmol/l for the raw carrot meal and 0·031 (sd 0·011) μmol/l for the stir-fried meal (P < 0·005), while for the carrot-derived material m β was found to be 0·011 (sd 0·004) μmol/l and 0·035 (sd 0·006) μmol/l for the raw and stir-fried meals, respectively (P = 0·03).
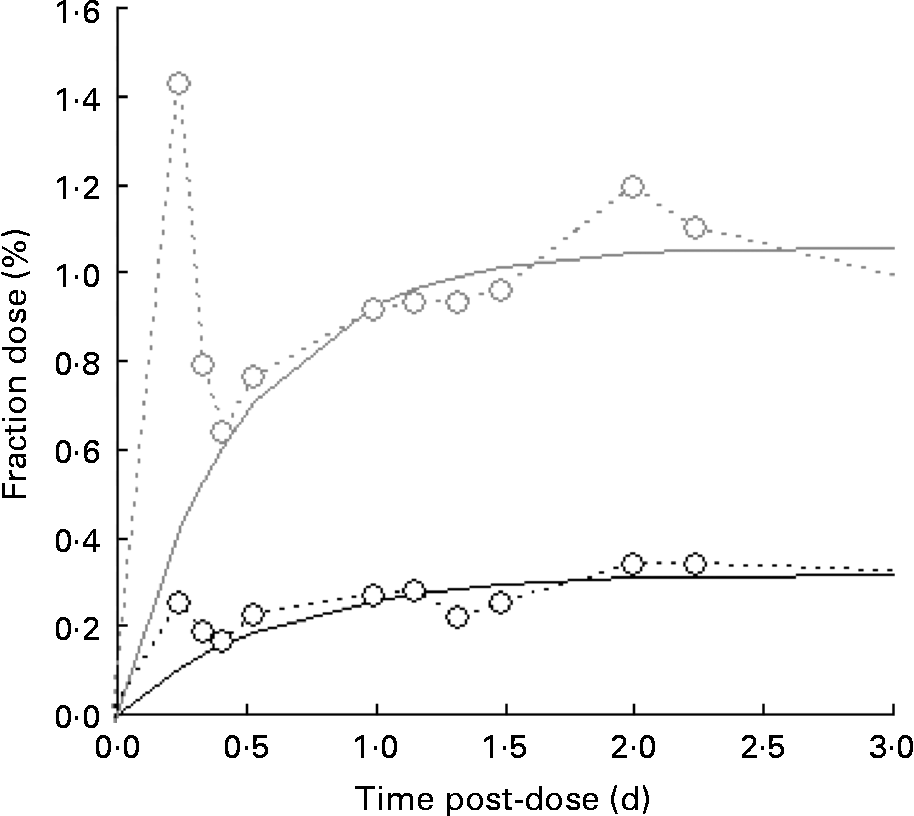
Fig. 4 Typical time course of plasma β-carotene, fraction of dose v. time profiles for the five subjects over a 4 d study period, after consumption of a single bolus meal of 100 g of the intrinsically labelled carrots (○; either raw or stir-fried in 10·5 ml groundnut oil), with 4·8 μmol [2H4]retinol acetate and 3·6 μmol [13C20]β-carotene in groundnut oil. Plasma C20 (![]() ) is derived from the [13C20]β-carotene dose.
) is derived from the [13C20]β-carotene dose.
Of more significance is the product of the two parameters, m βk β, since this represents the constant flux entering the sampled compartment. For [13C20]β-carotene, it was found to be 0·24 (sd 0·12) μmol/l per d for the raw carrot meal and 0·12 (sd 0·03) μmol/l per d for the stir-fried meal (not significantly different by the paired t test), while for the carrot-derived material, it was found to be 0·03 (sd 0·01) μmol/l per d and 0·14 (sd 0·01) μmol/l per d (P = 0·001) for the raw and stir-fried meals, respectively. Total β-carotene turnover can be derived by first multiplying the plasma volume by the concentration to give 1·2 (sd 0·2) μmol, which is in good agreement with Novotny(Reference Novotny, Dueker and Zech21), and then combining this with k β to obtain an estimate of turnover of 5·1 (sd 1·8) μmol/d.
Discussion
A comprehensive model of vitamin A metabolism has been given by Novotny(Reference Novotny, Dueker and Zech21) and is reproduced in Fig. 5. From this model, the qualitative aspects of the time profiles of plasma β-carotene and retinol following ingestion of a carotenoid-rich meal can be described (Fig. 6). Roughly 20 % of the ingested β-carotene is cleaved to retinol in the enterocytes, and both this and the parent compound appear transiently in the chylomicron lipids as they are transported to the liver. The liver acts as a buffer for both β-carotene and retinol, releasing both to the bloodstream in lipid fractions for transport to the sites of action and subsequent irreversible loss. The approximate half-life in plasma of the buffered β-carotene is predicted to be just over 3 d, with that of retinol being slightly lower at 2·8 d.

Fig. 5 Compartmental model of β-carotene metabolism in human subjects (adapted from Novotny et al. (Reference Novotny, Dueker and Zech21)). In this diagram, β-carotene is designated by rectangular boxes and retinol by ovals. The values above the arrows are fractional rate constants for inter-compartmental transfer deduced from the flows and masses given in the original.

Fig. 6 Qualitative aspects of the time profiles of plasma β-carotene and retinol following ingestion of a carotenoid-rich meal, containing 7 μmol β-carotene, according to the model given by Novotny et al. (Reference Novotny, Dueker and Zech21).
While this model qualitatively describes the kinetics of β-carotene and retinol that we observed in the present study, the model is too complicated to be tested by our measurements, and therefore we were unable to use it to quantitatively investigate our experimental data. Instead, we have used simple one- and two-compartment models to describe plasma β-carotene and retinal, respectively. Our models are not based on detailed physiological considerations, but instead are selected on a principle of parsimony so that they may be described with the minimum experimental data.
Unfortunately, an ambiguity is likely to arise when analysing data in terms of these models in the absence of other information. This is illustrated by considering the solution for a mammillary model, which is known to be:
where g 1 is the fractional rate constant of the input to, and g 2 that of the output from, the sampled compartment. However, simple rearrangement gives
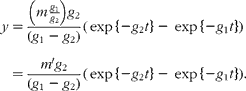
In other words, in the absence of further evidence, it is not possible to unambiguously assign the rate constants to influx or efflux. In the cases encountered in the present study, any decision must be based on how well the parameter m or m′ can be reconciled with the total flux of material through the pool.
The sizes of the doses administered in the present study were designed to mimic habitual daily intake. A total of just over 10 μmol β-carotene were ingested with the meal, which is in the range of typical daily intakes of 5–12 μmol(Reference Koushik, Hunter and Spiegelman22). In contrast, other stable isotope studies of vitamin A metabolism have used doses of pharmacological size(Reference Novotny, Dueker and Zech21, Reference Haskell23). The doses employed in the present study can be regarded as tracer in nature, since there was no observed perturbation of either the plasma β-carotene or retinol pools.
Modelling the kinetics of vitamin A metabolism is made difficult by hepatic take-up and re-release of both retinol itself and the provitamins such as β-carotene. When β-carotene is ingested, only a fraction is absorbed and transported to the bloodstream. During this process, some are converted to retinyl palmitate, and another fraction is irreversibly lost. Chylomicron β-carotene, therefore, does not represent the total vitamin A, which is derived from the dietary provitamin. Additionally, the rapid nature of its metabolism requires frequent blood sampling for its measurement. Lipoprotein β-carotene, on the other hand, is derived from a large buffer stored in the liver, and therefore an ingested bolus enjoys a prolonged re-release into the bloodstream. The kinetics are much slower, but there would have been more material lost by conversion to retinol in the liver. Conversely, chylomicron retinol reflects the fraction of dietary β-carotene, which is absorbed and converted in the enterocytes, while the other plasma retinol (bound to protein) is again released from the buffered hepatic stores.
In the present study, we chose to add extrinsically labelled material to our intrinsically labelled meal to act as reference standards. Our hypothesis is that a labelled β-carotene could be used to determine the bioavailability with respect to the free compound of that material in a food matrix, while labelled retinol would allow the partitioning of the provitamin to retinol to be investigated. As discussed earlier, the investigation of total vitamin A kinetics requires frequent blood sampling in the period immediately after dose administration followed by a period of relatively infrequent samples over a longer time period. However, in order to reduce the burden on our volunteers, we chose not to study the acute phase closely, but instead to focus on the longer-term kinetics, since these are more useful in assessing the amount of useful retinol ultimately derived from the meal.
Because of the nature of the way it was given, it was expected that the dose of retinol acetate would be completely and rapidly absorbed. The acute chylomicron response observed is not inconsistent with this assumption. In the kinetic curves, a maximum concentration is achieved very quickly (in < 6 h), followed by a slower decay back to basal levels over the next 24 h or so. This behaviour can be predicted by a simple model comprising a compartment with either rapid input and relatively slow output, or with slow input and relatively rapid output. We have chosen the former, and furthermore made the assumption that the rapid nature of the input is such that it can be approximated to a bolus. This reduces the model to that shown in Fig. 2(a), and allows the identification of the experimental parameter k R with the fractional rate of clearance of retinyl palmitate from plasma. This interpretation is supported by its identification of the parameter m R with the total amount of substrate to pass through the pool, assumed to be the plasma. The product of estimated plasma volume and m R amounts to 78 % of the dose of retinyl palmitate administered for the raw carrots and 70 % for the stir-fried ones, which is consistent with our hypothesis that the method of administration of retinol acetate would render it highly bioavailable.
Our choice of rapid uptake and relatively slow elimination of retinyl ester is discordant with the interpretation of Novotny et al. (Reference Novotny, Dueker and Zech21). In their interpretation with slow uptake and rapid elimination, the maximum instantaneous quantity of retinyl ester in the chylomicron pool given by:
is about 1·2 % of the dose given, which is incompatible with the observed maximum concentration. Transposing the assignment of the input and output rate constants given by Novotny predicts a maximum instantaneous quantity in the pool of 93 % of the dose approximately 0·75 h after dose administration, which is in good agreement with the present observations. We therefore identify our estimated value for k RE with that (erroneously) associated with retinyl ester absorption in the Novotny model.
Further evidence for the fast uptake/slow elimination model that we propose is the close agreement between our value for k 2 and its associated half-time of about 0·25 d and that found for chylomicron clearance itself(Reference Bitzen, Winqvist and Nilssonehle24). A lower limit can be placed on k in (by virtue of the maximum concentration must occur before 0·25 d) of 5·2/d, which is indicative of the ready bioavailability of retinol acetate in palm oil.
Protein-bound retinol derived from chylomicron-bound retinyl esters begins to appear in the plasma approximately 6 h after meal ingestion, after which it can be described by a two-compartment model. Again, there is a potential ambiguity with defining which of the two fractional rate constants obtained represents plasma clearance, which can be resolved by the plasma concentration. Assigning the rate constants g 1 = k 02 and g 2 = k 21, and assuming that the retinal-binding protein is confined to plasma (the minimum possible distribution volume), then the total quantity of [2H4]retinol to pass through this pool after the raw carrot meal is ![]() (10 % of the dose), whereas if the assignment is reversed, it is found to be about 160 % of the dose given. On this basis, the kinetics of hepatic release of protein-bound retinol into plasma is relatively fast (k 21 ≈ 10 per d), with relatively slow irreversible loss (k 21 ≈ 0·8 per d).
(10 % of the dose), whereas if the assignment is reversed, it is found to be about 160 % of the dose given. On this basis, the kinetics of hepatic release of protein-bound retinol into plasma is relatively fast (k 21 ≈ 10 per d), with relatively slow irreversible loss (k 21 ≈ 0·8 per d).
The fraction of the [2H4]retinyl ester dose, which is re-released into the plasma bound to protein, was not significantly different between the meals (10·7 (sd 0·5) % after the raw carrot meal and 11·4 (sd 1·7) % after the stir-fried meal). Combining this with the results from the retinyl ester kinetics indicates that about 15 % of the ‘absorbed’ retinol is bioavailable in the short-term. Presumably the remaining 85 % are sequestered in deeper hepatic stores.
The amount of dietary free β-carotene equivalent to 1 μmol of dietary retinyl ester is calculated by:
and found to be 2·9 (sd 0·8) for the raw carrot meal and 3·7 (sd 1·0) for the stir-fried meal (paired t test, P = 0·04). These values are of the same order as the generally accepted retinol equivalent (RE; 3·2 μmol β-carotene/μmol retinol, equivalent to 6 μg/μg), much higher than would be expected if β-carotene cleaved symmetrically and both halves of the molecule were utilised, which would give a value of 0·5 μmol/μmol.
The difference in [13C10]β-carotene behaviour between the meals is further illustrated by comparing m β. The amount of the tracer material found in plasma lipoprotein from the stir-fried meal was 56 (sd 13) % of that from the raw carrot meal. It therefore appears that the reduced free retinol equivalent from the stir-fried meal is due to poorer absorption of the free β-carotene, rather than differences in post-absorptive processing.
The relative bioavailability of β-carotene from the carrots and the free compound is calculated via
which was found to be 11 (sd 5) % for the raw carrot meal and 74 (sd 19) % for the stir-fried meal (P = 0·01). Finally, the RE of carrot-derived β-carotene can be calculated by combining the observation of retinol and β-carotene:
which is found to be 41 (sd 16) μmol/μmol for the raw carrots, but only 6·2 (sd 2·5) μmol/μmol when the carrots are stir-fried. Stir-frying therefore increases carotenoid bioavailability from the carrot more than six-fold.
The RE of β-carotene in food has been the subject of investigations over many years. Initial estimates of 6 μg carotene/μg retinol ( ≡ 3·2 μmol/μmol)(25–Reference Bieri and McKenna27) were derived from the work of Hume & Krebs(Reference Hume and Krebs28). This was based on the assumptions that retinol was derived from absorbed β-carotene on an equimolar basis, and that, on average, dietary β-carotene was about one-third as bioavailable as retinol. Subsequently, this value has been revised to 12 μg/μg ( ≡ 6·4 μmol/μmol) by the US Institute of Medicine(29). However, it is naive to imagine that a single RE is universally applicable to all foods – not only does the food itself matter but factors such as cooking and other processing methods have been shown to have a profound effect(Reference de Pee, West and Muhilal30–Reference Tang, Qin and Dolnikowski35). Recognition that these and other factors determine carotenoid bioavailability from foods has led to the introduction of the SLAMENGHI mnemonic(Reference de Pee and West36).
The RE values obtained in the present study (77 μg/μg for raw carrots and 11·6 μg/μg when stir-fried) fall each side of the value of 14·8 μg/μg reported for the steamed vegetable(Reference Tang, Qin and Dolnikowski35), and may be compared with that of 13 μg/μg for sautéed sweet potato, 10 μg/μg for steamed sautéed Indian spinach(Reference Haskell, Jamil and Hassan37) and 21 μg/μg(Reference Tang, Qin and Dolnikowski35) for steamed spinach. Clearly, cooking generally increases bioavailability, and from this limited evidence, the addition of oil or fat in frying is the optimal method of those investigated so far. However, the present study has merely demonstrated that two factors could have contributed to the difference in the yield of retinol: either the frying process or the use of more oil (10·5 ml) for stir-frying the carrots is effective in increasing bioavailability. It was the intention of the present study to compare raw carrot with stir-fried as might be performed in meal preparation in a typical family kitchen. The quantity of oil we chose was sufficient to cook the carrots without leaving appreciable amounts in the pan when the carrots were served. Investigations into the optimal quantity of oil are required in future. In summary, when formulating strategies for the use of vegetables as a sustainable and effective way of combating VAD, cooking methods must be considered.
Acknowledgements
We would like to acknowledge the Medical Research Council for funding the present study. We would like to thank Kerry Jones, Antony Wright, Neal Matthews and Glynn Harvey for their technical advice and knowledge regarding method development and instrumental analysis. We would also like to thank the volunteers for participating in the present study and Mario Siervo, Sarah Jones and Desda Jordan who assisted with the phlebotomy and volunteer study of this work. Finally, we would like to thank Ann Prentice, Susan Jebb, Gita Mishra, Mary Pennant, Sarah Casey, Adrian Izzard and Cheryl Kidney for their continued support and kind help. A. G., L. J. C. B. and W. A. C. contributed to the design of the study. A. G. performed the actual study including the meal design and preparation, volunteer work, HPLC and GC–MS analysis of the plasma and food, and data analysis. There are no conflicts of interest.
Appendix 1. Calculation of the enrichment of β-carotene from GC–MS data
The cracking pattern of the molecular cluster of perhydro β-carotene can be analysed by assuming that the molecule contains two species of hydrogen, one from the native carotenoid made up of fifty-six sites with fractional abundance F a, and the other added in the hydrogenation comprising twenty-two sites at abundance F b, in addition to the carbon with abundance F C. Elementary permutation and combination theory(Reference Bluck and Volmer13) gives the theoretical intensity of each isotopologue of the compound as:
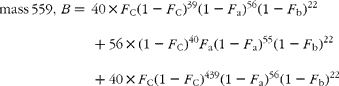
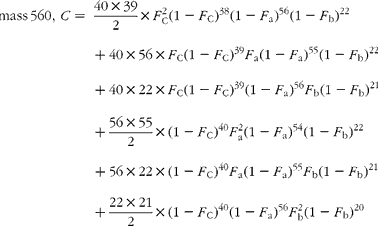

In GC–MS, there is no absolute measure of spectral intensity, and results are reported as ratios to the parent peak. Therefore,
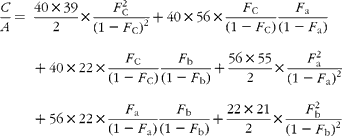
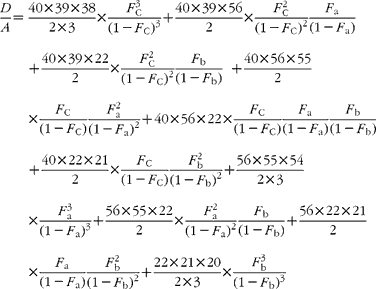
If carbon and b-hydrogens are at natural abundance (F C = 0·0111 and F b = 0·00 015)(Reference Bohlke, de Laeter and De Bievre14), then these become:
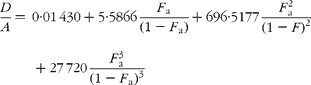
The best estimate of F a can now be obtained by least squares fitting to the experimentally observed mass spectral ratios.
Appendix 2. Calculation of β-carotene mole fractions in plasma
The contributions to the mass spectra in the range 558–580 m/z are summarized in Table 1. From this, it is possible to derive expressions for the six isotopomer ratios measured with respect to the peak at 558 m/z:
Simple rearrangement leads to the set of six simultaneous equations:
![\begin{eqnarray} \left. {\begin{array}{ccc}[( A _{U} - A _{Y}) R _{559} - ( B _{U} - B _{Y})] X _{Y} + ( A _{U} R _{559} - B _{U}) X _{C} = A _{U} R _{559} - B _{U} \\ [( A _{U} - A _{Y}) R _{560} - ( C _{U} - C _{Y})] X _{Y} + ( A _{U} R _{560} - C _{U}) X _{C} = A _{U} R _{560} - C _{U} \\ [( A _{U} - A _{Y}) R _{561} - ( D _{U} - D _{Y})] X _{Y} + ( A _{U} R _{561} - D _{U}) X _{C} = A _{U} R _{561} - D _{U} \\ ( A _{U} - A _{Y}) R _{578} X _{Y} + ( A _{U} R _{578} + A _{C}) X _{C} = A _{U} R _{578} \\ ( A _{U} - A _{Y}) R _{579} X _{Y} + ( A _{U} R _{579} + B _{C}) X _{C} = A _{U} R _{579} \\ ( A _{U} - A _{Y}) R _{580} X _{Y} + ( A _{U} R _{580} + C _{C}) X _{C} = A _{U} R _{580} \\ \end{array} }\right \}. \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU28.gif?pub-status=live)
In the matrix form, this is written as:
![\begin{eqnarray} \left [{\begin{array}{ccc}( A _{U} - A _{Y}) R _{559} - ( B _{U} - B _{Y}) & ( A _{U} R _{559} - B _{U}) \\ ( A _{U} - A _{Y}) R _{560} - ( C _{U} - C _{Y}) & ( A _{U} R _{560} - C _{U}) \\ ( A _{U} - A _{Y}) R _{561} - ( D _{U} - D _{Y}) & ( A _{U} R _{561} - D _{U}) \\ ( A _{U} - A _{Y}) R _{578} & ( A _{U} R _{578} + A _{C}) \\ ( A _{U} - A _{Y}) R _{579} & ( A _{U} R _{579} + B _{C}) \\ ( A _{U} - A _{Y}) R _{580} & ( A _{U} R _{580} + C _{C}) \\ \end{array} }\right ]\left [{\begin{array}{ccc} X _{Y} \\ X _{C} \\ \end{array} }\right ] = \left [{\begin{array}{ccc} A _{U} R _{559} - B _{U} \\ A _{U} R _{560} - C _{U} \\ A _{U} R _{561} - D _{U} \\ A _{U} R _{578} \\ A _{U} R _{579} \\ A _{U} R _{580} \\ \end{array} }\right ]. \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU29.gif?pub-status=live)
This is of the form:
which has the well-known least-squares solution:
Worked example
The mass spectrum obtained from a plasma sample is:
Previous measurements have indicated that the coefficients of spectrum of the pure components are
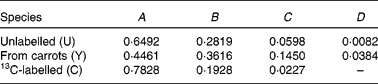
Therefore,
![\begin{eqnarray} { H _{\lowbar } } = \left [{\begin{array}{ccc}0\cdot 6492\times 0\cdot 44\,541 - 0\cdot 2819 \\ 0\cdot 6492\times 0\cdot 09\,881 - 0\cdot 0598 \\ 0\cdot 6492\times 0\cdot 01\,604 - 0\cdot 0082 \\ 0\cdot 6492\times 0\cdot 10\,940 \\ 0\cdot 6492\times 0\cdot 02\,692 \\ 0\cdot 6492\times 0\cdot 00\,331 \\ \end{array} }\right ] = \left [{\begin{array}{ccc}0\cdot 0073 \\ 0\cdot 0044 \\ 0\cdot 0022 \\ 0\cdot 0710 \\ 0\cdot 0175 \\ 0\cdot 0021 \\ \end{array} }\right ] \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU32.gif?pub-status=live)
and
![\begin{eqnarray} {\begin{array}{ccc}{ C _{ = } } & = \left [{\begin{array}{ccc}(0\cdot 6492 - 0\cdot 4461)\times 0\cdot 44\,541 - (0\cdot 2819 - 0\cdot 3616) & 0\cdot 6492\times 0\cdot 44\,541 - 0\cdot 2819 \\ (0\cdot 6492 - 0\cdot 4461)\times 0\cdot 09\,881 - (0\cdot 0598 - 0\cdot 1450) & 0\cdot 6492\times 0\cdot 09\,881 - 0\cdot 0598 \\ (0\cdot 6492 - 0\cdot 4461)\times 0\cdot 01\,604 - (0\cdot 0082 - 0\cdot 0384) & 0\cdot 6492\times 0\cdot 01\,604 - 0\cdot 0082 \\ (0\cdot 6492 - 0\cdot 4461)\times 0\cdot 10\,940 & 0\cdot 6492\times 0\cdot 10\,940 + 0\cdot 7828 \\ (0\cdot 6492 - 0\cdot 4461)\times 0\cdot 02\,692 & 0\cdot 6492\times 0\cdot 02\,692 + 0\cdot 1928 \\ (0\cdot 6492 - 0\cdot 4461)\times 0\cdot 00\,331 & 0\cdot 6492\times 0\cdot 00\,331 + 0\cdot 0227 \\ \end{array} }\right ] \\ & = \left [{\begin{array}{ccc}0\cdot 1702 & 0\cdot 0073 \\ 0\cdot 1053 & 0\cdot 0044 \\ 0\cdot 0334 & 0\cdot 0022 \\ 0\cdot 0222 & 0\cdot 8538 \\ 0\cdot 0055 & 0\cdot 2102 \\ 0\cdot 0007 & 0\cdot 0248 \\ \end{array} }\right ] \\ \end{array} }. \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU33.gif?pub-status=live)
Therefore,
and
The pseudo-inverse of ![]() is then
is then
![\begin{eqnarray} \left ({ C _{ = } }^{ T }{ C _{ = } }\right )^{ - 1}{ C _{ = } }^{ T } = \left [{\begin{array}{ccc}4\cdot 1392 & 2\cdot 5608 & 0\cdot 8133 & - 0\cdot 0474 & - 0\cdot 0118 & - 0\cdot 0008 \\ - 0\cdot 1078 & - 0\cdot 0669 & - 0\cdot 0202 & 1\cdot 1046 & 0\cdot 2720 & 0\cdot 0321 \\ \end{array} }\right ]. \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU36.gif?pub-status=live)
Finally,
Meaning that the β-carotene comprised 87·8 % pre-existing (unlabelled), 3·9 % from the carrots and 8·2 % from the 13C-labelled standard.
Appendix 3. Calculation of retinol mole fractions in plasma
The contributions to the mass spectra in the range 358–371 m/z are summarized in Table 2. From this, it is possible to derive expressions for the six isotopomer ratios measured with respect to the peak at 558 m/z:
Simple rearrangement leads to the set of eleven simultaneous equations:
![\begin{eqnarray} \left. {\begin{array}{ccc}[ B _{U} - B _{Y} - ( A _{U} - A _{Y}) R _{359}] X _{Y} + ( B _{U} - A _{U} R _{359}) X _{C} + ( B _{U} - A _{U} R _{359}) X _{D} = B _{U} - A _{U} R _{359} \\ [ C _{U} - C _{Y} - ( A _{U} - A _{Y}) R _{360}] X _{Y} + ( C _{U} - A _{U} R _{360}) X _{C} + ( C _{U} - A _{U} R _{360}) X _{D} = C _{U} - A _{U} R _{360} \\ [ D _{U} - D _{Y} - ( A _{U} - A _{Y}) R _{361}] X _{Y} + ( D _{U} - A _{U} R _{361}) X _{C} + ( D _{U} - A _{U} R _{361}) X _{D} = D _{U} - A _{U} R _{361} \\ ( A _{U} - A _{Y}) R _{362} X _{Y} + A _{U} R _{362} X _{C} + ( A _{D} + A _{U} R _{362}) X _{D} = A _{U} R _{362} \\ ( A _{U} - A _{Y}) R _{363} X _{Y} + A _{U} R _{363} X _{C} + ( B _{D} + A _{U} R _{363}) X _{D} = A _{U} R _{363} \\ ( A _{U} - A _{Y}) R _{364} X _{Y} + A _{U} R _{364} X _{C} + ( C _{D} + A _{U} R _{364}) X _{D} = A _{U} R _{364} \\ ( A _{U} - A _{Y}) R _{365} X _{Y} + A _{U} R _{365} X _{C} + ( D _{D} + A _{U} R _{365}) X _{D} = A _{U} R _{365} \\ ( A _{U} - A _{Y}) R _{368} X _{Y} + ( A _{C} + A _{U} R _{368}) X _{C} + A _{U} R _{368} X _{D} = A _{U} R _{368} \\ ( A _{U} - A _{Y}) R _{369} X _{Y} + ( B _{C} + A _{U} R _{369}) X _{C} + A _{U} R _{369} X _{D} = A _{U} R _{369} \\ ( A _{U} - A _{Y}) R _{370} X _{Y} + ( C _{C} + A _{U} R _{370}) X _{C} + A _{U} R _{370} X _{D} = A _{U} R _{370} \\ \left ( A _{U} - A _{Y}\right ) R _{371} X _{Y} + \left ( D _{C} + A _{U} R _{371}\right ) X _{C} + A _{U} R _{371} X _{D} = A _{U} R _{371} \\ \end{array} }\right \} \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20160319073618744-0991:S000711451100451X_eqnU49.gif?pub-status=live)
In the matrix form, this is written as
![\begin{eqnarray} \left [{\begin{array}{ccc}[ B _{U} - B _{Y} - ( A _{U} - A _{Y}) R _{359}] & ( B _{U} - A _{U} R _{359}) & ( B _{U} - A _{U} R _{359}) \\ [ C _{U} - C _{Y} - ( A _{U} - A _{Y}) R _{360}] & ( C _{U} - A _{U} R _{360}) & ( C _{U} - A _{U} R _{360}) \\ [ D _{U} - D _{Y} - ( A _{U} - A _{Y}) R _{361}] & ( D _{U} - A _{U} R _{361}) & ( D _{U} - A _{U} R _{361}) \\ ( A _{U} - A _{Y}) R _{362} & A _{U} R _{362} & ( A _{D} + A _{U} R _{362}) \\ ( A _{U} - A _{Y}) R _{363} & A _{U} R _{363} & ( B _{D} + A _{U} R _{363}) \\ ( A _{U} - A _{Y}) R _{364} & A _{U} R _{364} & ( C _{D} + A _{U} R _{364}) \\ ( A _{U} - A _{Y}) R _{365} & A _{U} R _{365} & ( D _{D} + A _{U} R _{365}) \\ ( A _{U} - A _{Y}) R _{368} & ( A _{C} + A _{U} R _{368}) & A _{U} R _{368} \\ ( A _{U} - A _{Y}) R _{369} & ( B _{C} + A _{U} R _{369}) & A _{U} R _{369} \\ ( A _{U} - A _{Y}) R _{370} & ( C _{C} + A _{U} R _{370}) & A _{U} R _{370} \\ ( A _{U} - A _{Y}) R _{371} & \left ( D _{C} + A _{U} R _{371}\right ) & A _{U} R _{371} \\ \end{array} }\right ]\left [{\begin{array}{ccc} X _{Y} \\ X _{C} \\ X _{D} \\ \end{array} }\right ] = \left [{\begin{array}{ccc} B _{U} - A _{U} R _{359} \\ C _{U} - A _{U} R _{360} \\ D _{U} - A _{U} R _{361} \\ A _{U} R _{362} \\ A _{U} R _{573} \\ A _{U} R _{364} \\ A _{U} R _{364} \\ A _{U} R _{365} \\ A _{U} R _{369} \\ A _{U} R _{370} \\ A _{U} R _{371} \\ \end{array} }\right ]. \end{eqnarray}](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary-alt:20160626131722-06715-mediumThumb-S000711451100451X_eqnU50.jpg?pub-status=live)
This is of the form:
which has the well-known least-squares solution:










