


Introduction
Though intracranial aneurysms (IAs) can occur sporadically, they are more common in individuals that have a family history of the disease, and on average, familial IAs develop at an earlier age.Reference Vlak, Algra and Brandenburg 1 , Reference Broderick, Brown and Sauerbeck 2 To date, several common variants have been statistically associated with IA through genome-wide association, candidate gene, and linkage studies.Reference Tromp, Weinsheimer and Ronkainen 3 As the next generation sequencing technology has become more accessible, research groups have been able to search for rare variants in exomes and explore the possible modes of inheritance for this disease in families with non-syndromic IA.Reference Farlow, Lin and Sauerbeck 4 , Reference Yan, Hitomi and Takenaka 5 However, no causative variants have been identified, and our knowledge of the genetic etiology of familial IA is still limited.
There appears to be a high prevalence of familial IA in the province of Newfoundland and Labrador (NL), Canada. Over a 5-year recruitment period, we ascertained 53 IA families containing 99 affected individuals. The unique population of NL has provided geneticists with a wealth of opportunity to study familial disease. The current population has grown from a group of initial immigrants of mainly Irish and English descent who settled in isolated communities around the island in the mid-1700 s.Reference Rahman, Jones and Curtis 6 This settlement has led to the relatively high prevalence of several monogenic disorders, including Bardet–Biedl syndrome, Lynch syndrome, and arrhythmogenic right ventricular cardiomyopathy.Reference Young, Penney and Woods 7 – Reference Merner, Hodgkinson and Haywood 9 The presence of familial IA in the population was first referenced in a case study of familial central nervous system disorders, which described three families with seven individuals that had been diagnosed with subarachnoid hemorrhage (SAH).Reference Maroun, Murray and Jacob 10 Previous studies have also reported an increased prevalence of familial IA in similar genetic isolates, such as the French-Canadian population of Québec and the Finnish population.Reference Zhou, Ambalavanan and Rochefort 11 – Reference Wills, Ronkainen and van der Voet 13
In order to elucidate the genetic risk factors involved with familial IA development in this province, we investigated the presence of shared, rare variants in affected family members from two large kindreds, through the use of whole exome sequencing (WES). It was predicted that one or more strongly penetrant variants would be present in each family. Pathogenic variants identified in this cohort could lead to the identification of causative genes and affected pathways in other populations.
Methods
Participant Recruitment and Phenotyping
The Health Research Ethics Board (Reference Number 04.89) for Memorial University of Newfoundland approved this study, and all participants provided informed consent. The overall study design for this pilot study is shown in Figure 1. Our cohort was assembled through collaboration between Memorial University’s Discipline of Genetics and the Department of Surgery (Division of Neurosurgery) at Eastern Health in St. John’s, NL, Canada, which is the only neurosurgical unit in the province. Patients were referred to our study if they had presented with either ruptured or unruptured IA. Diagnosis of IA was confirmed through computed tomography (CT) imaging of the Circle of Willis or by magnetic resonance (MR) angiography. Patients were asked whether they had a family history of IA, and additional family members were then contacted for participation. At the date of publication, 154 affected individuals and 415 unaffected family members had been consented to this study. Our database also includes 105 individuals with unknown affection status. Relatives of patients that provided consent were offered CT angiography, underwent a comprehensive medical and family history assessment, and a blood sample was drawn if possible. Genomic DNA was extracted from whole blood using either a simple salting out method or the Wizard® Genomic DNA Purification kit (Promega, Madison, WI, USA). For some participants, sufficient clinical data were obtained (age of diagnosis, rupture status, etc.), but molecular investigations could not be pursued. These circumstances included individuals who were deceased prior to participant recruitment, who were consented by proxy by a relative, so that we could use their clinical data as part of our larger study. As well, participants living outside the province that did not arrange to have blood provided were not included in the genetic portion of our study.
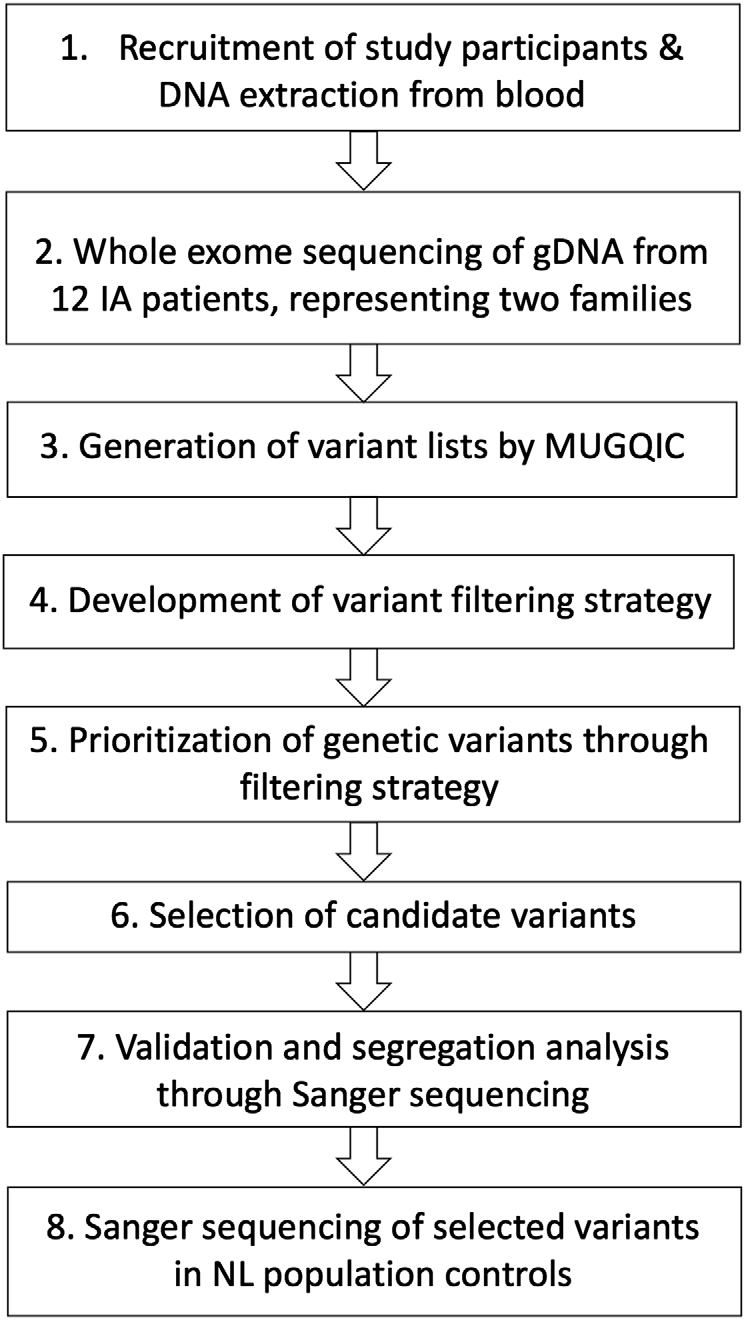
Figure 1: Study design for genetic investigations. Abbreviations: genomic DNA (gDNA), Newfoundland and Labrador (NL), McGill University and Genome Quebec Innovation Centre (MUGQIC).
For all affected participants, histories encompassed as much detailed information as possible such as age at diagnosis, number of IAs, dimensions and location of any IAs, rupture incidence as well as any courses of treatment provided. Information regarding two known risk factors, cigarette smoking and hypertension, was also recorded. Affected participants were ultimately placed into one of the three categories: familial (≥2 first-degree relatives with IA), sporadic (no known family history), or equivocal (not enough data to categorize). Our cohort includes over 50 families with possible shared genetic risk factors. Eight families contained four or more affected individuals.
Selection of Study Families
For this pilot study, we chose to focus on two of the families that have a particularly strong family history of IA, specifically eight or more first-degree relatives with IA. Each family has more than 11 affected family members in total, either living or deceased. Eligible affected participants for our pilot study must have had (1) a diagnosis of unruptured IA through CT or MR imaging, or a history of ruptured IA or SAH and (2) at least one affected relative (first or second degree) with IA. Exclusion criteria included (1) individuals that did not provide informed consent, (2) individuals that did not provide a blood sample for genetic analysis, and (3) individuals under the age of 18. Phenotype characteristics for the 20 affected individuals that met these criteria are provided in Table 1.
Table 1: Phenotype characteristics for affected study participants from families R1256 and R1352
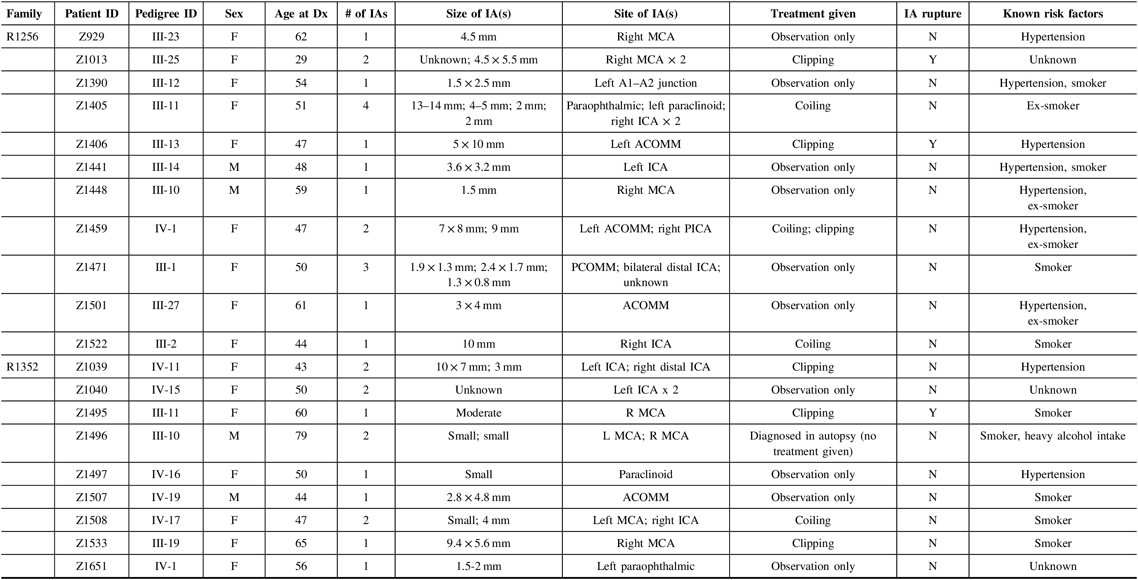
F = female; M = male; ICA = internal carotid artery; ACA = anterior carotid artery; MCA = middle cerebral artery; PCOMM = posterior communicating artery; PCA = posterior cerebral artery; ACOMM = anterior communicating artery; Dx, diagnosis; Y = yes; N = no; mm = millimeters.
Figure 2 illustrates condensed pedigrees for these two multiplex families. A younger generation beyond the four shown has been documented but was eliminated from our molecular research, as children were not recruited into the study. For the purpose of this study, we acted under the assumption that familial IA reflected an unknown monogenic inheritance pattern, with the knowledge that additional risk factors play a role in disease development and rupture. Likewise, due to IA’s variable expressivity and penetrance, unaffected family members were also not included in initial molecular analyses. They were however used to test the segregation of candidate variants.
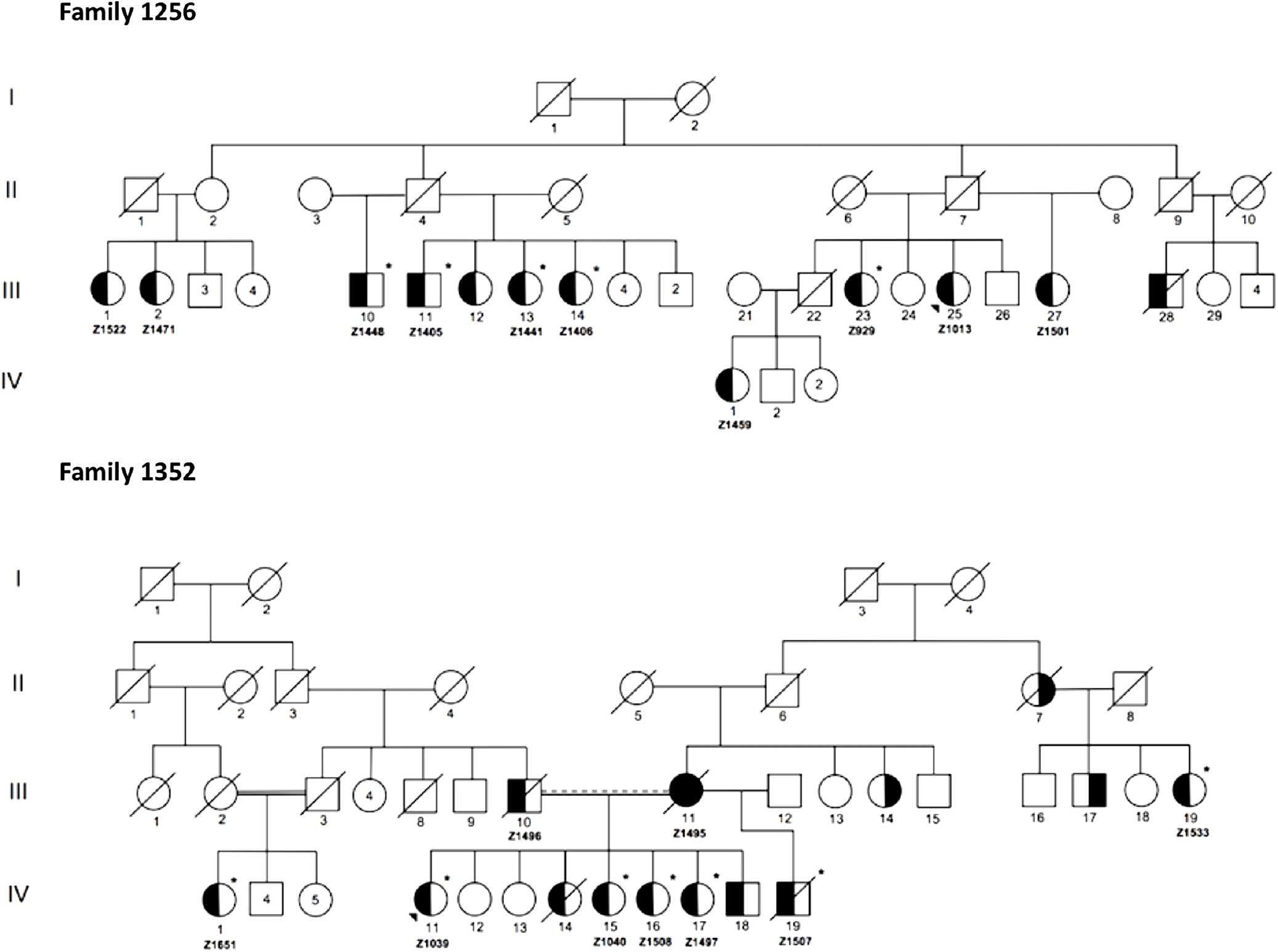
Figure 2: Condensed pedigrees for R1256 and R1352. Study numbers starting with “Z” are located under each affected individual who consented to participate. III-1 and II-5 of R1352 represent the same individual. Dotted marriage line indicates possible consanguinity. Symbols: squares (males), circles (females), left side shaded (IA diagnosis), right side shaded (AAA diagnosis), unshaded (unaffected or phenotype unknown), asterisk (exome sequenced), diagonal line (known to be deceased), shaded triangle (proband).
WES Analysis
From the two families, a total of 12 affected individuals were selected for WES, 5 from R1256 and 7 from R1352 (Figure 2). As shown in Table 1, the additional eight affected individuals were excluded from WES as in some cases it was determined through quality control that there was no sufficient high-quality DNA needed for robust sequencing. As well, we had limitations with regard to available personnel and resources, which limited our study size. WES was completed at McGill University and Genome Quebec Innovation Center (MUGQIC, Montreal, QC, Canada). The Agilent SureSelect 50 Mb All Exon kit was used for exome capture and paired-end sequencing was completed on the Illumina® HiSeq 2000 platform. Following sequencing, initial bioinformatics analyses were completed at MUGQIC using an in-house pipeline for alignment, variant calling, and annotation. Further details are provided in the online supplementary methods.
Variant Filtering Strategy
A filtering strategy was developed to eliminate any variants that were unlikely to play a role in IA pathogenesis. First, annotated variant lists for each family were grouped by predicted impact. For the purpose of our analyses, we filtered each type but focused on variants of high and moderate effect, which excluded synonymous variation from consideration (low impact). High-impact variants included splice-site acceptor, splice-site donor, start-lost, frameshift, stop-gain, stop-lost, and rare amino acid substitution types, while moderate-impact variants were classified as missense variants, according to the provided categorization from MUGQIC. Next, variants with a read depth less than 10× were eliminated. Variants were then prioritized if they were shared by the majority of affected individuals in at least one family. Variants were filtered out if they were present in less than 4/5 exomes in R1256 or less than 6/7 exomes in R1352. Common variants were eliminated next, through the use of population frequency data from the MUGQIC in-house control exome set, and the following data sets: dbSNP, ExAC Browser, NHLBI Exome Variant Server, and Ensembl Genome Browser.Reference Smigielski, Sirotkin and Ward 14 – Reference Cunningham, Amode and Barrell 17 Variants with a minor allele frequency greater than or equal to 1% in any of these databases were eliminated. Following this step, there were still a high number of moderate-effect variants remaining for family R1256. To suit the parameters and constraints of this pilot project, an additional step was added to focus solely on variants found in 5/5 exomes in this family that were also unreported in variant databases. We modeled the disease inheritance as both recessive and dominant when filtering the variants in both families.
Validation and Segregation in Families
Sanger sequencing was employed to validate filtered variants in affected individuals and eliminate any false positive findings. In this step, an additional five affected family members from R1256 were included, for whom sufficient DNA was available (Z1522, Z1471, Z1459, Z1013, and Z1501 in Figure 2). Primer sequences and specific protocols for polymerase chain reaction are available upon request.
After a list of top candidates was created, additional measures of prioritization were applied. Several statistical algorithms for predicting variant pathogenicity were employed, specifically PolyPhen2, sorting intolerant from tolerant (SIFT), and genomic evolutionary rate profiling, which gives a measure of evolutionary conservation of an amino acid.Reference Adzhubei, Schmidt and Peshkin 18 – Reference Cooper, Stone and Asimenos 20 All top candidates were then reviewed manually to ensure that filtering steps were applied properly and that no obviously benign or common variants were remaining. Top candidates were sequenced in a selection of 100 population control samples from NL. These control DNA samples were from NL residents who were recruited through random-digit dialing as part of the Newfoundland Colorectal Cancer Registry project.Reference Wang, Dicks and Gong 21
Results
Summary of Results
After consolidating our clinical data for the 20 participants in this study, we were able to generate some basic statistics for our cohort. This led into the successful completion of WES, which provided us with 12 exomes for analysis. Through our variant filtering strategy, 6 genetic variants of interest were prioritized in family R1352, and 17 were prioritized in family R1256. It was determined that there were no shared variants between the two families. Segregation analysis with Sanger sequencing showed that the missense variant SPDYE4 c.C103T (p.P35S) was present in 10 of the 11 affected individuals from family R1256 and was absent in 100 control samples from the NL population. This led us to categorize it as a top candidate in this family. The high-impact variant C4orf6 c.A1G (p.M1V) was present in six of seven affected individuals and was absent in 100 control samples as well in family R1352. The fact that this variant was classified as high impact led us to categorize it as a top candidate in the family.
Phenotyping of Study Cohort
Based on the data collected in Table 1, we were able to provide a detailed phenotype for all 20 affected participants in our study. In the process, several observations were made, and characteristics of the cohort were defined. For the 20 affected study participants across both families, the average age of diagnosis was 52.3, while the age range of initial diagnosis was 29–79. The 79-year-old male (Z1495) from family R1352 was an outlier in our set, who was found to have two small IAs during autopsy. Our cohort consists of 16 females (80%) and 4 males (20%). Nine individuals from our cohort had been previously diagnosed with hypertension, and 13 had cigarette smoking (previously or currently) as a risk factor. Five of these individuals had both hypertension and a smoking-related history. These two risk factors were not disclosed for three individuals (Z1013, Z1040, and Z1651). As well, eight individuals were reported as having more than one IA, all of whom were females. There was no clear pattern across each family in terms of aneurysm size and/or location, as diagnoses were quite variable.
WES and Variant Filtering
After WES, a range of 40–63 million reads were generated for each library in our set of 12 exomes. The average coverage for the whole genome was 2.8% (total number of aligned reads/size of the genome) and the ratio of surviving reads following trimming to the number of raw reads was 91.85% across all 12 exomes.
Following the application of our stringent filtering strategy, the number of surviving variants was greatly reduced in each family. Figure 3 outlines the number of variants remaining after each filtering step. After step four, only 6 variants in family R1352 (1 high impact and 5 moderate impact), and 68 variants in family R1256 (2 high impact and 66 moderate impact) were prioritized for further investigation (see Supplementary Tables 1 and 2). The 66 moderate-impact variants were filtered further and reduced to a more manageable list of 15 that were present in 5 of 5 exomes and were apparently novel according to our variant annotation, leaving us with 17 variants to investigate in this family (see Supplementary Table 2).
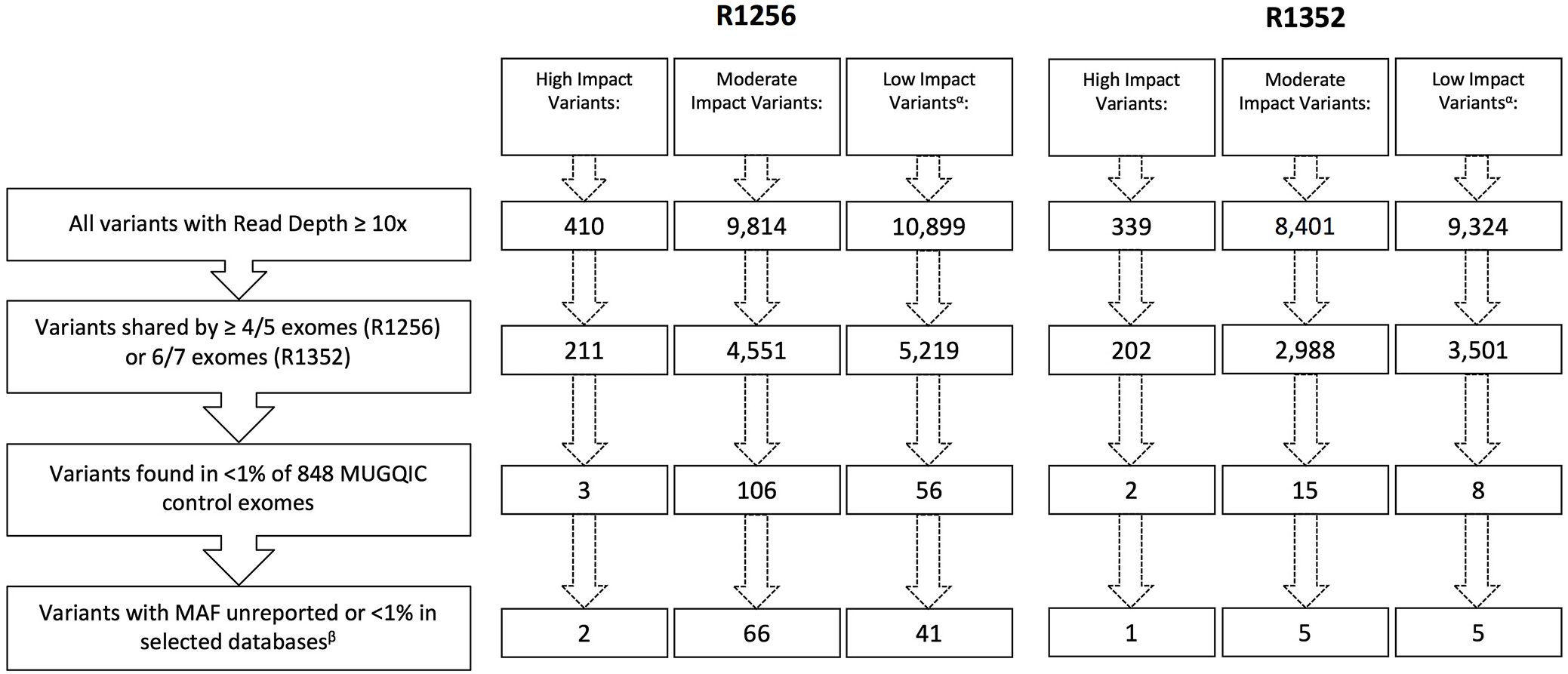
Figure 3: Remaining variants in each impact category following application of filtering strategy. αLow-impact variants were excluded from further analyses beyond filtering. βSelected databases include dbSNP, ExAC Browser, Ensembl Genome Browser, and NHLBI Exome Variant Server.
Validation and Segregation
Following Sanger sequencing validation and elimination of false positives, it was acknowledged that there were no shared variants between the two families. As well, there were no variants in the same gene across both families.
In family R1256, two high-impact variants were confirmed: a frameshift variant in CCDC3 and a splice-site variant in OCIAD1 (Table 2). For this family, we were fortunate to have a high recruitment rate for affected individuals and their DNA. Therefore, we were able to sequence these variants in additional affected relatives. CCDC3 c.425delA (p. Tyr267ThrfsTer21) was only validated in 5 of 10 sequenced individuals, though it had been detected in six exomes. This could have been an error in variant calling and was flagged for further review. OCIAD1 c.G-6+1A was present in six of eight individuals that were successfully sequenced for this difficult region of the gene.
Table 2: Variants remaining following the application of filtering strategy and segregation testing by Sanger sequencing in family R1256 exomes
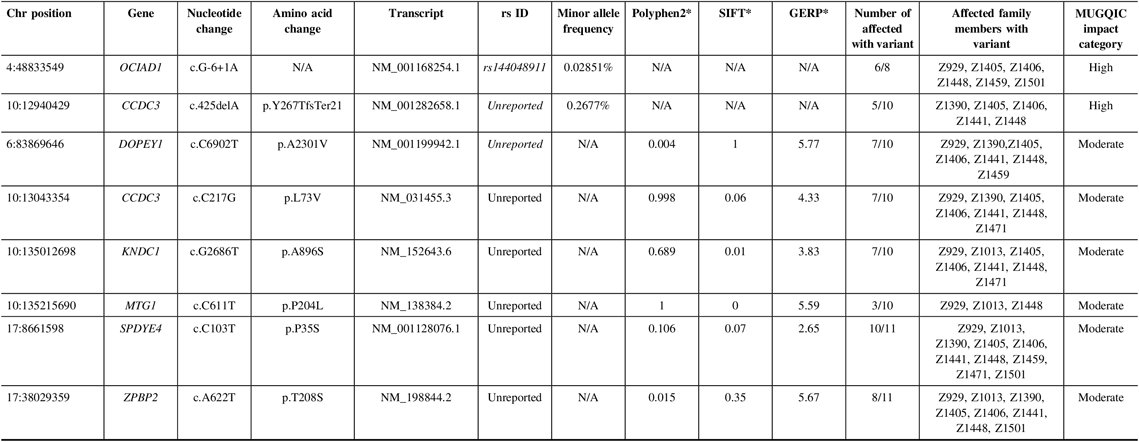
N/A = not applicable; Het = heterozygous; Hom = homozygous; MUGQIC = McGill University and Genome Quebec Innovation Centre; chr = chromosome; SIFT = sorting intolerant from tolerant; GERP = genomic evolutionary rate profiling.
* PolyPhen2 scores range from 0 (benign) to 1 (damaging), SIFT scores range from 0 (damaging) to 1 (benign), and GERP scores range from −12.3 to 6.17, where 6.17 is the most conserved (Adzhubei et al.Reference Adzhubei, Schmidt and Peshkin18; Kumar et al.Reference Kumar, Henikoff and Ng19; Cooper et al.Reference Cooper, Stone and Asimenos20; Exome Variant Server 16).
Of the 15 moderate impact variants prioritized, validation of only six was successful (Table 2), as the remainder were located in a highly polymorphic region of the exome. Missense variants in DOPEY1, CCDC3, and KNDC1 were confirmed in 7 of 10 affected individuals. MTG1 c.C611T (p.P204L) was found in only 3 of 10, possibly due to sequencing or read depth quality. We were able to successfully obtain sequence from family member Z1522 to test the two remaining variants. Therefore, we were able to validate ZPBP2 c.A622T (p.T208S) in 8 of 11 affected individuals, and SPDYE4 c.C103T (p.P35S) in 10 of 11. This variant had the highest degree of segregation with IA incidence, compared to the other candidates. Following sequencing in unaffected relatives, 8 of 14 individuals had the SPDYE4 variant. Again, this step allowed us to develop a better picture of the variant segregation but was not used as a filtering step to exclude variants. Sanger sequencing of 100 population controls also revealed an absence of the SPDYE4 variant, and it was identified as the top candidate for IA susceptibility in this data set.
In family R1352, a single high-impact variant, C4orf6 c.A1G (p.M1V), passed the filtering criteria and was confirmed to be a true variant following Sanger sequencing; it was present in six of seven exomes (Table 3). Additional sequencing determined that 5 out of 14 selected unaffected relatives were also carriers of this variant. This variant had conflicting results with regard to in silico predictive tools, as it was reported to be benign by PolyPhen2 and deleterious by SIFT. As this was the only high-impact candidate remaining in the family, it was chosen to be Sanger sequenced in the NL population control data set. C4orf6 c.A1G (p.M1V) was not found in any of the 100 control samples and was identified as a top candidate in this family.
Table 3: Variants remaining following the application of filtering strategy and segregation testing by Sanger sequencing in family R1352 exomes
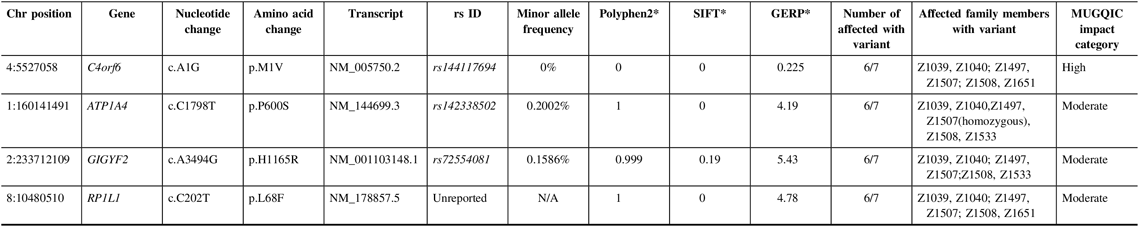
N/A = not applicable; Het = heterozygous; Hom = homozygous; MUGQIC = McGill University and Genome Quebec Innovation Centre; chr = chromosome; SIFT = sorting intolerant from tolerant; GERP = genomic evolutionary rate profiling.
* PolyPhen2 scores range from 0 (benign) to 1 (damaging), SIFT scores range from 0 (damaging) to 1 (benign), and GERP scores range from −12.3 to 6.17, where 6.17 is the most conserved (Adzhubei et al.Reference Adzhubei, Schmidt and Peshkin18; Kumar et al.Reference Kumar, Henikoff and Ng19 ; Cooper et al.Reference Cooper, Stone and Asimenos20; Exome Variant Server16).
Additionally, validation proved that three missense variants in ATP1A4, GIGYF2, and RP1L1 were true positives and were present in six out of seven exomes (Table 3). Genetic function was explored for all three variants, using databases such as UniProt and GeneCards.Reference Stelzer, Rosen and Plaschkes 22 , 23 All three variants were present in 4–5 unaffected relatives; therefore, the results of sequencing these individuals did not add any value to variant prioritization.
Discussion
It is apparent that IA is a genetic disorder in the featured multiplex families, due to the number of closely related affected individuals and the presence of IA in multiple generations. Other analyses using this abundance of phenotype data could be beneficial and could be used to rank phenotype severity. For example, family members with larger aneurysms or rupture history could have additional environmental or genetic risk factors compared to individuals with smaller IA dimensions. The risk factors of hypertension and smoking could also be included in the filtering process, and further investigation into these factors in the unaffected relatives could provide insight into the complexity of this disease.
The use of a geographically isolated population provides several advantages to our study. It is widely known that the NL population is distributed throughout a collection of outports and that families and neighbors are proficient at maintaining information about their genealogy. Isolated populations also have a more consistent genetic landscape due to low rates of migration and a reduced number of disease-causing alleles due to the presence of a genetic bottleneck. They also have a high tendency to participate in research, which is shown through the strong recruitment to our study. Clinical data and active participation from families prove invaluable when completing molecular investigations in a complex disease background. In light of the fact that the genetic landscape of NL is isolated and unique, the variants detected in this study may not be detected at the same allele frequencies in other populations.Reference Kristiansson, Naukkarinen and Peltonen 24
During the course of this pilot study, manuscripts from Farlow et al. and Yan et al. were released, which detailed the use of WES in families affected by IA. Each provided a list of prioritized variants for further investigation. The data from these studies was compared to our variant lists, and there was no overlap across these three studies.Reference Farlow, Lin and Sauerbeck 4 , Reference Yan, Hitomi and Takenaka 5
With the aid of next generation sequencing technology, we were able to identify and prioritize rare variants in our own set of 12 exomes. Following validation, it was confirmed that neither variant from our filtered list was detected in all affected participants from a single family. However, the design of our filtering strategy provided some room for incomplete segregation. As it is known that IA can occur sporadically, and is heavily influenced by lifestyle factors, we chose to examine variants that were detected in all exomes but also variants that were found in four of five exomes from R1256 and six of seven exomes from R1352. The presence of phenocopies could account for the lack of a shared variant in affected individuals. Unruptured IAs have a prevalence of approximately 3.2% and thus occur relatively commonly in the general population.Reference Vlak, Algra and Brandenburg 1 This prevalence could be elevated in our isolated and homogenous population, and it is possible that some family members with strong environmental risk factors could have a separate cause entirely.Reference Kristiansson, Naukkarinen and Peltonen 24
Though incomplete segregation was present, further functional investigations could be pursued to determine the biological implications of the C4orf6 and SPDYE4 variants. Currently, there is limited information about the function of these genes. At this point in our project, we have concluded that it is unlikely that a single rare variant is responsible for predisposition to IA in our cohort from NL. Through our preliminary genetic investigations in this cohort, several new questions have been stimulated with regard to the genetic etiology of familial IA. Through the interpretation of results and structure of the pedigree in family R1352, we considered the possibility of digenic inheritance in this disease. For example, in this family, four siblings (Z1039, Z1040, Z1497, and Z1508) and one half-sibling (Z1507) shared both C4orf6 c.A1G (p.M1V) and GIGYF2 c.A3494G (p.H1165R). Individual Z1533 had the GIGYF2 variant, while Z1651 had the C4orf6 variant. There was no clear connection between IA severity (size and rupture) and the combination of these two variants. However, it is an area that could be pursued further as digenic or polygenic inheritance has been largely unexplored in familial IA studies. As well, it is likely that there is genetic heterogeneity in this disease that will result in various causative IA genes throughout our group of families.
There were several limitations to our study, which can be improved upon in future analyses of our unique cohort. In any exome sequencing project, there are technical limitations to consider. Coverage of the exome can be impacted by DNA quality and stringent quality control surrounding read depth. It is also possible that genetic factors influencing IA development are found outside of the exome entirely. As our knowledge of the non-coding region of the genome increases, we will be able to confidently pursue whole genome-wide methods of analysis in the future. The filtering strategy that we designed for this study could also be a limitation. It is possible that our methods were too stringent for this cohort and the characteristics of the disease. It is possible that variants involved in IA predisposition are not as universally rare as we predicted and may have a global minor allele frequency higher than 1% and are thus less penetrant in general. As well, there are gaps in the available data that hindered our ability to interpret and prioritize variants. It is possible that some of the genes that we prioritized could have IA-related functionality that is yet unknown.
Overall, this study provided a starting point for further study of familial IA in a unique genetic isolate. In conjunction with researchers that are pursuing WES analysis in other populations, we hope to add to the growing body of knowledge surrounding this disease. The identification of any common biological pathways or gene categories prioritized through different WES studies could be a significant step forward.
Acknowledgments
We would like to thank all of the study participants and their families, without whom this research would not be possible. We would like to acknowledge Krista Mahoney for her assistance with preliminary data collection as well as Carol Negrijn and Marian Crowley for their assistance in acquiring clinical information.
Funding
This project was primarily funded by the Heart and Stroke Foundation of Canada (Grant Number: G-15-0008943), and a grant from the Medical Research Fund at Memorial University’s Faculty of Medicine.
Disclosures
None.
Statement of Authorship
AEP assembled the manuscript, created and applied the variant filtering strategy to exome data, and completed Sanger sequencing. BAF and FM collaborated in the creation of this study and are responsible for patient recruitment and phenotyping, along with the assistance of BN. FM also coordinated and performed screening and diagnosis of individuals. MOW directed the design of the molecular genetics study, supervised data collection and analysis, and assisted in primary manuscript revisions. All authors approved the final manuscript.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2019.230.








