


INTRODUCTION
Rift Valley fever virus (RVFV) (family Bunyaviridae, genus Phlebovirus) is a mosquito-borne virus which causes large outbreaks of disease in domestic ruminants such as sheep, cattle and goats in Africa whenever exceptionally heavy rains favour the breeding of the mosquito vectors [Reference Swanepoel, Coetzer, Coetzer and Tustin1]. Infection is usually benign in humans, and although only a small proportion of patients (0·5–1%) develop fatal encephalitis or haemorrhagic fever there may be many casualties in the course of large outbreaks [Reference Swanepoel, Coetzer, Coetzer and Tustin1]. The recent extension of RVFV to Saudi Arabia and Yemen, the fact that mosquitoes capable of transmitting the virus are widely distributed, the potential effects of global warming on climate, plus fears of bioterrorism, have raised international concern that the disease could spread to new regions, and emphasizes the need for safe and effective vaccines [Reference Swanepoel, Coetzer, Coetzer and Tustin1–Reference Sidwell and Smee4].
Smithburn modified live virus (SMLV) vaccine prepared from a mouse neuroadapted strain of RVFV, has been used extensively for immunization of livestock in Africa, but is less effective in cattle than in sheep and is only partially attenuated, causing abortion or teratology in a proportion of pregnant animals [Reference Swanepoel, Coetzer, Coetzer and Tustin1, Reference Smithburn5]. There is concern that SMLV could revert to full virulence, and hence its use is contraindicated in countries where RVFV is not known to be endemic [Reference Swanepoel, Coetzer, Coetzer and Tustin1]. Formalin-inactivated vaccine is more expensive to produce, and requires multiple initial inoculations and regular boosters to induce effective immunity [Reference Swanepoel, Coetzer, Coetzer and Tustin1, Reference Barnard6]. Consequently, it is less useful in livestock than modified live virus, particularly when there is a need to attain rapid immunization in the face of an outbreak, and it has not found extensive application. An inactivated vaccine was used successfully to protect laboratory and veterinary workers at risk of infection, but was released on a limited trial basis only [Reference Swanepoel, Coetzer, Coetzer and Tustin1, Reference Meadors, Gibbs and Peters7, Reference Pittman8]. A mutagen-derived strain of RVFV, MP12, which was developed as a candidate attenuated vaccine for use in both humans and livestock, produced encouraging results in initial trials in sheep and cattle, but may cause teratology when administered early in pregnancy [Reference Meadors, Gibbs and Peters7, Reference Hunter, Erasmus and Vorster9, Reference Morrill10].
Alternatives for production of RVF vaccines include alphavirus replicons: single-hit expression vectors in which a gene of interest replaces viral structural genes from a highly efficient internal subgenomic promoter. Replicon vector RNA is packaged by providing the viral structural proteins in trans, with the resulting replicon particles being capable of infecting target cells and expressing the heterologous genes to high levels. However, due to the lack of encoded homologous structural genes, replicon vectors are incapable of producing progeny virions [Reference Frolov11]. Replicons based on alphaviruses such as Venezuelan equine encephalitis virus (VEE), Semliki Forest virus (SFV) and Sindbis virus (SIN), have shown promise as vaccines against several pathogens, including Marburg haemorrhagic fever and Lassa fever viruses [Reference Hevey12, Reference Pushko13].
RVFV particles have spherical symmetry with a host cell-derived lipid envelope through which virus-coded glycoprotein spikes or peplomers project. The particles have a tripartite single-stranded RNA genome, with negative-sense polarity large (L) and medium (M), and an ambisense small (S), RNA segments each contained in a separate nucleocapsid [Reference Nichol, Knipe and Howley14, Reference Schmaljohn, Hooper, Knipe and Howley15]. The M segment RNA encodes a non-structural protein NSm of unknown function, plus a polyprotein precursor of the Gn and Gc glycoproteins which form the envelope peplomers that mediate binding and entry of virus into permissive cells [Reference Schmaljohn, Hooper, Knipe and Howley15, Reference Swanepoel16]. The glycoproteins induce production of neutralizing antibody and protective immunity against infection with RVFV [Reference Francis and Magill17–Reference Schmaljohn19].
Given the need for improved vaccines against RVFV, we have developed replicon vectors based on mosquito (AR86) and human (Girdwood) isolates of SIN from South Africa [Reference Malherbe, Strickland-Cholmley and Jackson20, Reference Weinbren, Kokernot and Smithburn21], expressing RVFV Gn and Gc glycoproteins plus NSm protein. Evaluation of these vectors demonstrated their ability to elicit RVFV-specific immune responses in a mouse model that were protective against systemic and mucosal RVFV challenge. These vectors also elicited RVFV neutralizing antibody responses in vaccinated sheep, suggesting that further evaluation of alphavirus replicon-based RVFV vaccines is merited.
METHODS
Replicon production
The tripartite helper system for the AR86-based replicon system has been described previously [Reference Heise, Simpson and Johnston22], where the replicon RNA, which lacks the viral structural genes and is therefore incapable of producing viral particles, is packaged by providing two defective helper RNAs encoding either the viral capsid protein or the viral glycoproteins. For the Girdwood replicon system, the non-structural coding region of the genome from nucleotides 41–6463 was transferred from the full- length infectious clone pG100 [Reference Suthar23] into the AR86-based replicon pREP89 [Reference Heise, Simpson and Johnston22], using restriction enzymes MfeI and BstBI. All non-structural coding differences between AR86 and GirdwoodS.A. reside within this region. The glycoprotein helper pGIRDGLY was constructed by transferring nucleotides 8584–11 268 from the glycoprotein-coding region of pG100 into the AR86 helper pHelp102 [Reference Heise, Simpson and Johnston22] using restriction enzymes StuI and EcoRI. The resulting construct encoded viral E3, E2, 6K, and E1 proteins identical to those of Girdwood virus. Since there are no differences in the capsid-coding regions of AR86 and Girdwood, the AR86 capsid helper, pCAP86 [Reference Heise, Simpson and Johnston22], was used for Girdwood replicon particle packaging.
The RVFV M segment RNA of the Egyptian ZH-501 strain of RVFV [Reference Collett24], kindly provided by Dr Michael Parker of the U.S. Army Medical Research Institute for Infectious Diseases, Frederick, MD (USAMRIID), was introduced into the pRgird replicon plasmid at a ClaI site at position 7668, which is immediately 3′ of the Girdwood 26S promoter 5′-UTR, to create the plasmid pRgird-RVFV(M). The RVFV M segment was also introduced into the AR86 replicon plasmid pREP91, using the same conserved ClaI site to create the plasmid pREP91-RVFV(M). In addition, the RVFV glycoprotein-coding sequence was introduced into the VEE- based replicon pVR21 to produce VRP-RVF(M), as described previously [Reference Filone25], and this replicon was used for making serological antigen. The RVFV M segment contains five in-frame start codons, the first four of which can be used to initiate translation of both Gn and Gc [Reference Schmaljohn19]. Furthermore, initiation of translation from the fourth methionine codon has been reported to enhance expression of both Gn and Gc [Reference Schmaljohn19], therefore, unlike the SIN replicons, which expressed the entire RVFV M segment, the pVR21-RVF construct started at the fourth methionine codon of the RVFV M segment.
Several control replicons were also generated. The enhanced green fluorescent protein (Egfp; Clontech, Palo Alto, CA, USA), was introduced into the ClaI site of pRgird at position 7668 to create the plasmid pRgird-Egfp. The Egfp-expressing AR86 replicon, pREP91-Egfp, has been described previously [Reference Heise, Simpson and Johnston22]. An additional irrelevant control replicon was constructed by introducing the HA protein from influenza strain A/PR/8 [Reference Winter, Fields and Brownlee26] into the ClaI site at position 7668 of pREP91. Control VEE replicons, VRP-SARS(S), were generated by introducing the S glycoprotein from the Urbani strain of the SARS coronavirus behind the 26S promoter of the pVR21 replicon [Reference Deming27]. The resulting constructs were sequenced to confirm their identities and ensure that their coding sequences were correct.
The pRgird and pREP91 replicon plasmids, as well as the two helper plasmids pCAP86 and pGIRDGLY, were linearized using PmeI, while all VEE replicons and helpers were linearized with NotI, for use as in vitro transcription templates. For SIN-derived replicons, transcripts for the replicons and the capsid and glycoprotein helpers were generated using mMessage mMachine SP6 kits (Ambion, Austin, TX, USA) according to the manufacturer's instructions. For Girdwood replicon packaging, two transcription reactions of replicon RNA were pooled with two transcription reactions of pGIRDGLY and pCAP86-derived RNA, while AR86 replicons were pooled with pHELP102 and pCAP86 RNA. VEE replicon and helper transcripts were generated using mMessage mMachine T7 transcription kits (Ambion) as described previously [Reference Balasuriya28].
The pooled transcripts were electroporated into BHK-21 cells (ATCC) as described previously [Reference Heise, Simpson and Johnston22]. In brief, BHK-21 cells, which were maintained in α-MEM supplemented with 10% donor calf serum (DCS), 10% tryptose phosphate broth (TPO4), and 0·29 mg/ml l-glutamine, where passaged once in medium where 10% fetal calf serum (FCS) replaced the 10% DCS. At the time of electroporation, the cells were dissociated with trypsin and washed once with RNase-free AccuGene PBS (Cambrex, Rockland, ME, USA), resuspended in aliquots of 1·2×107 cells in 0·7 ml RNAse-free PBS, mixed with the pooled RNA transcripts and transferred to 0·4 cm electroporation cuvettes (Invitrogen, Carlsbad, CA, USA). The cells were pulsed three times at 2-s intervals on a Bio-Rad Genepulser II electroporator (Bio-Rad, Hercules, CA, USA) set at 0·85 kV and 25 μF. Electroporated cells were transferred to 150 mm dishes in 25 ml α-MEM supplemented with 10% FCS, 10% TPO4, l-glutamine and incubated at 37°C, 5% CO2 for 24 h. The supernatant was harvested and 10% screened for the presence of replication-competent virus by two passages in BHK-21 cells which were monitored for virus-induced cytopathic effect (CPE) [Reference Balasuriya28]. Replicon stocks which exhibited no CPE within these test monolayers on the second passage were considered to be free of detectable replication-competent virus and were then cleared for use. At this point, the replicon stocks were concentrated by centrifugation over a 20% sucrose cushion in PBS, resuspended in PBS supplemented with 1% FCS, and stored at −80°C in 100 μl aliquots.
The titre of each replicon stock was assessed by inoculation of tenfold dilutions onto BHK-21 cells in 8-well chamber slides (LabTek Inc., Bloomington, IN, USA) which were incubated for 18 h, fixed with acetone for 5 min at 4°C, and stained with polyclonal anti-RVFV hyperimmune mouse ascitic fluid (RVFV HAF) (generously provided by Dr M. Parker, USAMRIID) at 1:800 dilution, followed by 1:200 biotinylated horse anti-mouse IgG antibody (Vector Labs, Burlingame, CA, USA) and Texas Red-labelled streptavidin (Jackson ImmunoResearch, West Grove, PA, USA). Antigen-positive cells were counted on a Nikon TE300 fluorescent microscope.
Replicon-derived RVFV protein expression was analysed by infecting BHK-21 cells in 6-well plates with REP91-RVF(M) or Rgird-RVFV(M) replicons at a multiplicity of infection (MOI) of 0·5. At 24 h post-infection, cells were lysed in NP40 buffer, the lysates separated on 8% SDS–PAGE gels, transferred to PVDF Immunoblot membrane (Bio-Rad) and probed for Western blot analysis with RVFV HAF diluted 1:2000. Alternatively, blots were probed with monoclonal antibodies directed against Gn (RV5 3G2-1A) or Gc (RVII 7B2-2-2) (generously provided by Dr George Ludwig, USAMRIID, Ft. Detrick, Frederick, MD, USA). Anti-Gc only worked in Western blot applications when protein was analysed under non-denaturing conditions and did not recognize a secreted form of Gn, while anti-Gn worked under standard denaturing conditions and recognized a secreted form of Gn (data not shown). In addition, BHK-21 cells in 8-well chamber slides (LabTek Inc.) were infected and examined by immunfluorescence as described above.
Antibody tests
RVFV glycoprotein antigen for the enzyme-linked immunosorbent assay (ELISA) was prepared by infecting Vero-E6 cells (ATTC no. CRL-1586) with VRP-RVF(M) at a MOI of 5·0. Cultures were maintained for 24 h in serum free Optimem (Gibco, Gaithersburg, MD, USA), washed three times with PBS, the cells lysed with NP40 lysis buffer for 5 min at 4°C, the lysate clarified by centrifugation, and stored in single-use aliquots at −80°C. Negative control antigen was produced by infecting Vero cells in the same manner with VRP-SARS(S).
ELISAs were performed in 96-well Immunolon 4HBX plates (Thermolab Systems, Franklin, MA, USA), and the optimal working dilution of VRP-RVF antigen (1:150) was determined in doubling dilution chessboard titration with RVFV HAF. Unless specified otherwise, reagent volumes were 100 μl, the diluent and washing fluid was PBS (pH 7·4), and incubations were performed for 1 h at room temperature (22°C). Wells in test plates were coated with antigen diluted in carbonate buffer (pH 9·6), absorbed overnight at 4°C, washed once, and post-coated with 0·2 ml volumes of 5% milk in PBS (pH 7·4). Sera from immunized mice were tested in half-log dilution steps from 1:50 (150, 500, 1500, etc.). Following incubation, wells were washed three times and bound antibody was detected with horseradish peroxidase-conjugated goat anti-mouse IgG (Southern Biotech, Birmingham, AL, USA). After further incubation and washing, O-phenylenediame dihydrochloride substrate (Sigma, St Louis, MO, USA) was added, allowed to react for 30 min, and the optical densities (OD) which were recorded at 450 nm, were plotted against the log of the serum dilution. A sigmoidal curve was fitted to the data using least-squares regression (Sigma Plot, Jendel Scientific, San Francisco, CA, USA), with the equation for the curve solved for log dilutions at which the OD450 value equalled an arbitrary cut-off value of 0·2. Sera were tested in parallel against control antigen prepared from cells infected with VRP-SARS(S) replicon.
Antigen slides for indirect immunofluorescence (IF) tests were prepared from Vero76 cells infected with RVFV VRL688/78, isolated from a cow in Zimbabwe in 1978, and tests performed as described previously [Reference Hunter, Erasmus and Vorster9, Reference Swanepoel16]. Sera were screened for IgG antibody activity at a dilution of 1:8 using fluorescein-labelled anti-mouse immunoglobulin (Cappel, Organon Teknika nv, Turnhout, Belgium), and end points determined by testing doubling dilutions of serum.
For virus neutralization (VN) tests, stock virus consisted of supernatant fluids from Vero cell cultures infected with RVFV VRL688/78 and stored at −70°C, and the tests were performed in microplates as described previously [Reference Swanepoel29].
Immunization and RVFV challenge experiments
In initial experiments, groups of 6-week-old C57B1/6 strain mice were inoculated into a footpad with 105 cell culture infectious units (IU) of pREP91-RVF(M) or pRgird-RVF(M) replicon in 10 μl PBS with 1% DCS. Control vaccinated mice received pREP91-HA. The mice were boosted with the same dose of replicon 4 weeks post-primary inoculation, exsanguinated 3 weeks later, and antibody responses were assessed by ELISA, IF and VN tests. Protection tests were performed in a biosafety level 3 (BSL3) facility using outbred NIH Swiss mice. Groups of 4-week-old mice were inoculated into a footpad with 105 IU of pRgird-RVF(M), REP91-RVF(M), or REP91-HA replicons in PBS, and boosted 4 weeks post-primary inoculation. Three weeks later groups of five mice were challenged with 100 mouse LD50 of RVFV VRL 688/78, via either the intraperitoneal or intranasal route, and a subset of the mice exsanguinated to be tested for antibody by IF and VN techniques. Challenged mice were monitored for illness and mortality over a period of 2 weeks. Additional mice immunized with pRgird-RVF(M) were kept for up to 8 months and groups of five challenged intraperitoneally at monthly intervals to determine the duration of the protective immune response. All animal studies were conducted in accordance with animal care and use policies at either the University of North Carolina or the National Institute for Communicable Diseases.
For sheep immunization studies, two cross-bred Dorper ewes were inoculated subcutaneously with 2·5×107 cell culture infectious doses of pRgird-RVF(M) replicon in 1 ml PBS. A third sheep was mock-vaccinated with pREP91-HA. The three sheep were boosted 4 weeks later with the same doses of replicon. Rectal temperatures were recorded twice daily for 14 days after inoculation, and blood collected weekly for 7 weeks to test for neutralizing antibody. The vaccinated sheep exhibited no adverse reactions following vaccine administration.
RESULTS
Characterization of expressed proteins
The RVFV Gn and Gc glycoproteins are the major targets of protective immunity [Reference Francis and Magill17–Reference Schmaljohn19], therefore, the RVFV M segment, which encodes the Gn and Gc glycoproteins, as well as the non-structural protein nsM, was introduced behind the 26S promoter of the SIN-based replicons, as outlined in Figure 1. Correct orientation of the RVFV M segment was confirmed by restriction digest and by sequencing the full-length replicon/RVFV M segment constructs. These studies initially tested replicon vectors based on two different, but closely related SIN isolates, AR86 [Reference Heise, Simpson and Johnston22, Reference Heise, Simpson and Johnston30, Reference Simpson31] and GirdwoodS.A. [Reference Malherbe, Strickland-Cholmley and Jackson20, Reference Suthar23]. AR86 is mouse adapted [Reference Heise, Simpson and Johnston22, Reference Heise, Simpson and Johnston30, Reference Simpson31], while GirdwoodS.A. is derived from an infected human and is less mouse adapted [Reference Malherbe, Strickland-Cholmley and Jackson20, Reference Suthar23]. Therefore we were interested in determining whether either of the vectors provided an advantage with respect to eliciting an effective anti-RVFV response. However, both replicons exhibited similar levels of heterologous protein expression (data not shown), and elicited similar levels of anti-RVFV antibody in vaccinated animals (Table 1), suggesting that at least in the mouse model, there is no measurable difference in effectiveness between these two vectors. Expression of RVFV glycoprotein from replicon-infected BHK-21 cells was confirmed by fluorescent staining with RVFV-HAF (Fig. 2 a). To confirm that the replicon-derived RVFV glycoproteins were of the correct size and properly processed, lysates from REP91-RVF(M)-infected BHK-21 cells were analyzed by SDS–PAGE and Western blot with RVFV-specific antibody. RVFV HAF specifically recognized a band in the 55–57 kDa range which corresponds to the predicted size of the RVFV GN and GC glycoproteins (Fig. 2 b), and the presence of both GN and GC was confirmed by Western blot with monoclonal antibodies against GN and GC. As shown in Figure 2 c, an antibody against GN specifically recognized a band of ~50 kDa in size, which is consistent with mature GN. The GC-specific antibody, which does not recognize a secreted version of GN (data not shown), only worked in Western blot applications under denaturing conditions. However, as shown in Figure 2 c, this antibody also specifically reacted with the RVFV antigen, indicating that both GN and GC are expressed from these SIN replicon vectors.
Fig. 1. Schematic representation of Sindbis virus replicon REP91-RVF(M) expressing Rift Valley fever virus M segment RNA, or control replicon REP91-HA expressing the HA gene from influenza strain A/PR/8.
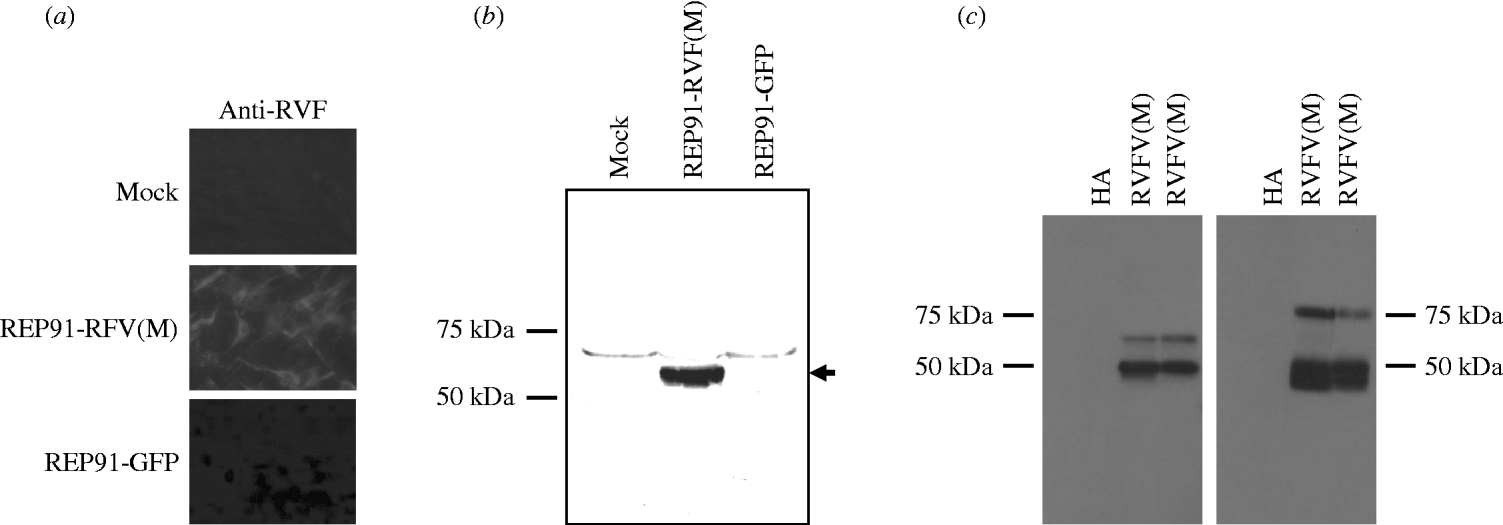
Fig. 2. Rift Valley fever virus (RVFV) glycoprotein expression by Sindbis virus replicons incorporating the RVFV M segment RNA. (a) BHK-21 cells infected with REP91-RVF(M) and stained by indirect immunofluorescence with anti-RVFV hyperimmune mouse ascitic fluid (RVF HAF). No staining was observed with control serum or in cells infected with the control REP91-HA. (b, c) BHK cells were infected with replicons expressing the RVFV M segment or control replicons expressing GFP (b) or influenza HA (c) and lysates probed by Western blot for RVFV glycoprotein expression with polyclonal (b) anti-RVFV HMAF or monoclonal antibodies (c) against Gn (left panel) or Gc (right panel). The RVFV(M) lanes in panel (c) represent duplicate samples.
Table 1. Antibody titres detected in mice 3 weeks post-boost with Sindbis virus-based replicons expressing Rift Valley fever glycoproteins

ND, Not done.
Immune responses in mice and sheep
To test whether the SIN-derived replicons were capable of eliciting RVFV-specific immune response, 6-week-old C57Bl/6 mice were inoculated subcutaneously in the left rear footpad with 105 IU replicon-expressing RVF(M), while control animals were mock-vaccinated. Mice were subsequently boosted with an additional 105 IU of the same replicon at 4 weeks post-primary inoculation, and serum anti-RVFV antibody responses evaluated by ELISA. As shown in Table 1, C57Bl/6 mice which had been given two doses of replicon pRgird-RVF(M) into the footpad produced RVF ELISA IgG antibody titres of 6266–61 085, IF titres of 2048–16 384, and neutralizing titres of 8–128, while mice mock-immunized with REP91-HA-expressing influenza haemagglutinin consistently tested negative for antibody to RVFV. Similar anti-RVFV responses were obtained in C57Bl/6 mice immunized with REP91-RVF(M) (data not shown). In ELISA assays, serum from immunized mice or RVFV HAF showed no reactivity with the control SARS-CoV S glycoprotein antigen.
In follow-up studies, outbred NIH Swiss mice were inoculated with replicons expressing the RVFV M segment, or influenza HA as a control. Mice vaccinated with either REP-91-RVFV(M) or Rgird-RVFV(M) developed IF titres of 2048–8192 and VN titres of 4–16, which were similar to the titres developed by the C57Bl/6 mice 3 weeks after immunization, while mice which had been mock-immunized with REP91-HA remained RVF antibody free (Table 1). Three weeks after immunization, groups of five mice immunized with REP91-RVF(M) or pGIRD-RVF(M) resisted challenge with RVFV 688/78 by either intraperitoneal or intranasal routes, while mock-immunized mice all succumbed by day 4 after intraperitoneal challenge, or day 5 after intranasal challenge (Table 2). Additional mice, which had been immunized with pRgird-RVF(M) and were challenged intraperitoneally with RVFV 688/78 in groups of five at monthly intervals for up to 8 months post-immunization, all survived without overt signs of illness (data not shown).
Table 2. Protection against lethal RVFV challenge

Although the mouse model is valuable for studying vaccine-induced protection against RVFV, sheep are highly susceptible to RVFV infection, and represent one of the most affected species during RVFV outbreaks. Therefore, studies were performed to test whether alphavirus replicons would induce RVFV-specific immunity in vaccinated sheep. In a preliminary study, two adult sheep received 2·5×107 IU Rgird-RVFV(M), while a single control animal received HA replicon as a control. Neutralizing antibody activity to RVFV was first detected at a titre of 1:4 in the serum of one sheep a week after the administration of the primary immunizing dose of replicon pRgird-RVF(M), while the second sheep developed a neutralizing titre of 1:4 at 2 weeks post-primary inoculation. By three weeks post-boost (week 7 post-primary inoculation) when monitoring was discontinued, the first sheep had a VN titre of 1:128 and the second a titre of 1:32. No virus-neutralizing antibody response to RVFV was detected in the control sheep immunized with pREP91-HA.
DISCUSSION
Control of RVFV in endemic areas can be achieved through annual inoculation of young livestock with vaccine which induces durable immunity. The existing SMLV vaccine could be used for the purpose since it is attenuated for non-pregnant animals and confers lifelong immunity. However, it has proved difficult to sustain immunization programmes during the irregular intervals of years between outbreaks, and consequently there is almost invariably indiscriminate and belated use of the abortigenic SMLV vaccine during epidemics following heavy rains. A separate economic burden arises from the banning of importation of livestock from countries where RVFV occurs, and in this respect the lack of distinction between antibody response induced by immunization and response to natural infection, constitutes a further disadvantage of present vaccines [Reference Swanepoel, Coetzer, Coetzer and Tustin1]. Therefore, we were prompted to examine the feasibility of developing an alphavirus replicon vaccine which would eliminate several safety concerns associated with the live attenuated vaccine and also permit vaccinated and naturally infected animals to be easily differentiated.
The VEE vector has been commonly used for alphavirus replicon candidate vaccines developed for use in humans, but veterinary application of this vector is unlikely to be accepted readily in regions from which VEE virus is absent. We opted to use SIN vectors since the virus is widely distributed in Africa where RVFV occurs, and antibody to the virus has been found in livestock in the absence of evidence of a corresponding disease [Reference McIntosh32, Reference Taylor33]. Therefore, although the use of tripartite helper systems decreases the chances that replication-competent recombinant viruses will be generated during replicon packaging and post-production screening is designed to detect any replication-competent virus that does arise, any recombinant virus arising during the replicon packaging process is likely to be attenuated and would at worst case simply be a mildly pathogenic SIN. Our findings demonstrated that SIN-based replicon vectors can express RVFV glycoproteins efficiently and elicit protective immune responses against lethal RVFV challenge in mice. This contrasts with recent work by Gorchakov et al. [Reference Gorchakov34] which suggested that SIN-based vectors were poor inducers of protective anti-RVFV immunity, while VEE vectors exhibited superior performance. However, the work by Gorchakov and colleagues involved a different vector and vaccination regimen, and vectors were derived from a different SIN strain, making direct comparisons between these two studies difficult. However, it does suggest that different SIN-based vector systems are likely to exhibit differences in efficacy and direct comparisons between these systems would be informative.
A promising aspect of the replicon vectors is their ability to elicit anti-RVFV antibody responses in sheep, and this suggests that alphavirus vectors could be used as a veterinary vaccine against RVFV. A potential advantage of a vectored vaccine would be the ability to differentiate immunized from naturally infected livestock on the basis of a lack of antibody response to RVFV nucleocapsid protein in vaccinated animals. Alternatively, unique immunological tags could be inserted into replicon vaccines. SIN-based replicon vaccine against RVFV could also find application in humans engaged in the livestock industry who are at occupational risk of exposure to infection. The disease is not considered to be contagious, and livestock generally acquire infection from transmission by arthropod vectors. In contrast, humans commonly become infected from contact with blood or other tissues of infected animals, but on occasion there appears to have been aerosol or droplet infection via mucosal surfaces, a phenomenon which has been demonstrated experimentally and which renders RVFV a candidate for use as a bioweapon [Reference Sidwell and Smee4, Reference Francis and Magill17, Reference Brown, Dominik and Morrissey35–Reference Miller38]. Hence, the apparent ability of SIN-based replicon vaccine to protect against intranasal RVFV challenge could be an important advantage.
Clearly, further investigation of SIN-based replicon vaccine against RVFV is warranted, with a need to establish the correlates of immunity to challenge more exactly, including determination of protective levels of neutralizing antibody, the potential role of non-neutralizing antibodies, plus the contributions of cell-mediated and mucosal immune responses [Reference Anderson39–Reference Thompson41]. Ultimately, a vaccine requiring only a single immunization would be optimal, especially for livestock applications. Preliminary results in vaccinated sheep indicate that neutralization titres of 1:4 are achieved 1–2 weeks post-primary vaccination, however, it is currently unknown whether these levels are sufficient to protect from RVFV challenge, or whether durable and protective anti-RVFV immunity can be achieved with only a single replicon immunization in either mice or sheep. Therefore, additional studies are needed to address these questions, and if necessary, replicon constructs could be modified to enhance the protection conferred on livestock by a single dose of vaccine. Given that Sindbis viruses are endemic to many of the same areas where RVFV occurs, it will also be important to determine whether pre-existing immunity to SIN [Reference McIntosh32, Reference Taylor33] has the capacity to limit efficacy of SIN replicon-based vaccine, and if so, develop strategies to overcome this problem.
ACKNOWLEDGEMENTS
This work was supported by a Developmental Award from the Southeastern Regional Center for Excellence in Biodefense and Emerging infectious diseases (SERCEB) 5 U54 AI057157-02 to M.T.H. and an award to F.J.B. by the Polio Research Foundation. We thank Martha Collier, Robert Johnston, and Nancy Davis at the Carolina Vaccine Institute for assistance in packaging the VEE-based replicons and Bianca Trollinger for assistance with Western blots and Sindbis virus replicon production. We also thank the members of the Carolina Vaccine Institute for helpful scientific discussions and critical reading of the manuscript.
DECLARATION OF INTEREST
None.




