


INTRODUCTION
Japanese encephalitis virus (JEV), an important member of the Flaviviridae family, can cause severe viral encephalitis in humans. JEV is prevalent in the temperate and tropical areas of eastern and southern Asia. Affected countries include China, Japan, Korea, Philippines, Vietnam, Cambodia, Indonesia, Laos, Malaysia, Thailand, India, Nepal, Sri Lanka, Bangladesh and Bhutan [Reference van den Hurk1, Reference Erlanger2]. The virus is transmitted by mosquitoes, principally of the Culex species. Pigs are necessary for pre-epizootic amplification of the virus [Reference van den Hurk1]. The virus exists in a zoonotic cycle between mosquitoes and pigs (and/or water birds) and humans are dead-end hosts [Reference van den Hurk1]. About 30 000–50 000 cases of JEV infection with 10 000 deaths are reported annually [Reference Tsai3].
JEV contains a single-stranded positive-sense RNA genome, which is about 11 kb in length. The genome contains a single, long open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTR). The ORF is translated into a large polyprotein, which is processed into three structural proteins (C, M, E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) [Reference Chambers4, Reference Sumiyoshi5]. Based on the nucleotide sequences of the envelope (E) gene, five genotypes have been identified [Reference Mohammed6–Reference Chen9]. In recent years, considerable efforts have been made to characterize the genetic diversity and origins of JEV, using sequencing and phylogenetic analysis of complete envelope or genome sequences from strains newly isolated [Reference Huang10–Reference Li19]. However, there have been few attempts to elucidate the molecular evolution of JEV.
China is representative of countries that experience annual JEV transmission. Since the first report in 1949, JEV has circulated in China for over 60 years. Except for Xinjiang and Qinghai provinces, all other provinces have reported cases of JEV infection. Historically, JEV prevalence was very high in 1960s and early 1970s and the most serious outbreaks occurred in 1966 and 1971, with a reported disease incidence of over 20/100 000 people [Reference Gao20]. After the nationwide vaccination programme initiated in 1970s, JEV-infected cases decreased markedly, with disease incidence declining from 20·92/100 000 in 1971 to 0·23/100 000 in 2008 [Reference Gao20]. However, 7860, 5422, 5097, 7643, 4660, 2975, 3913, and 2738 cases were still reported from 2003 to 2010, respectively (www.chinacdc.cn).
In this study, we analysed all JEV strains which were isolated in China from 1949 to 2009 in order to elucidate the epidemiological patterns and molecular evolution of JEV.
METHODS
Sequences
All JEV envelope gene sequences from 161 strains isolated in China (from 1949 to 2009) were collected from GenBank as of 10 August 2011 (www.ncbi.nlm.nih.gov). The following criteria were used when choosing the sequences: (1) all sequences had a known date of isolation; (2) all sequences containing stop codons were excluded; (3) sequences with 100% similarity were excluded. No recombinant strains were identified in these cohorts. All sequences and alignments in this study are available from authors upon request.
Phylogenetic analysis
The TREE-PUZZLE program [Reference Schmidt21] was used to infer maximum-likelihood trees for JEV. The tree was rooted by using West Nile virus strain NY99 (DQ211652).
Molecular evolution analysis
The rate of nucleotide substitution per site and the time to the most recent common ancestor (TMRCA) were estimated using the Bayesian Markov Chain Monte Carlo (MCMC) approach as implemented in the BEAST 1.5.3 package (http://beast.bio.ed.ac.uk) [Reference Drummond22]. The analysis was performed by using the GTR+G+Г4 substitution model under a coalescent model of constant population size. In each case, the relaxed (uncorrelated lognormal) molecular clock model was used (the relaxed clock model was favoured over the strict clock model in these datasets). A final chain length of 10 million or 20 million was employed to make an effective sample size (ESS) for parameter estimates >200. The resulting convergence was analysed by using Tracer 1.4.1 (http://evolve.zoo.ox.ac.uk). To reveal uncertainty in the estimations, 95% high probability density (HPD) intervals in each case was also determined.
Selection pressures between genotypes
Overall selection pressures acting on the envelope gene of each dataset were determined as the ratio of non-synonymous (d N) to synonymous (d S) substitutions (d N/d S) per site by using the pair-wise method of Nei & Gojobori as implemented in MEGA4 [Reference Tamura23]. To identify the positive selection sites in the envelope gene of each dataset, the Datamonkey facility [Reference Pond24] was employed and the single-likelihood ancestor counting (SLAC), fixed-effects likelihood (FEL), internal FEL (IFEL), and random-effects likelihood (REL) methods were used (http://www.datamonkey.org).
RESULTS
Phylogenetic analysis
All JEV strains isolated in China fell into three genotypes I, III, and V (Fig. 1). All strains which were grouped into genotype I, were isolated in the mainland after 2000 except for three strains (YN83-Meng83-54, YN79-Bao83, YN86-86266) isolated in Yunnan prior to 1987 and two strains (YL0806f, TPC0806C) isolated in Taiwan in 2008. Genotype III was divided into two clades. One clade (named ‘Taiwan clade’) only included strains isolated in Taiwan. However, the other clade (named ‘mainland clade’) consisted of strains from the mainland (71/81) except for minor strains (10/81) which were isolated mainly in Taiwan. As described previously [Reference Li25], XZ0934 fell into genotype V.

Fig. 1. JEV strains in China, in genotypes I, III, and V.
Co-circulation of genotypes I and III
As shown in Figure 2, the JEV strains used in this study covered 17 provinces in China (34 provinces in total) including Heilongjiang, Liaoning, Beijing, Shanxi, Henan, Hubei, Sichuan, Yunnan, Guizhou, Guangxi, Shandong, Jiangsu, Zhejiang, Shanghai, Fujian, Taiwan, and Tibet. Co-circulation of genotypes I and III was confirmed in nine of the provinces, which included Liaoning, Shanxi, Henan, Sichuan, Yunnan, Guizhou, Shanghai, Zhejiang, and Taiwan. Only genotype III was confirmed as circulating in Fujian, Heilongjiang, Beijing, Shandong, Jiangsu, and Hubei. In Guangxi province, only genotype I was confirmed as circulating. Genotypes I and V co-circulated in Tibet. Table 1 shows the numbers of isolates in genotypes I and III in each endemic province.

Fig. 2. Map of China showing provinces where JEV strains were circulating.
Table 1. Numbers of isolates in genotypes I and III in each endemic province
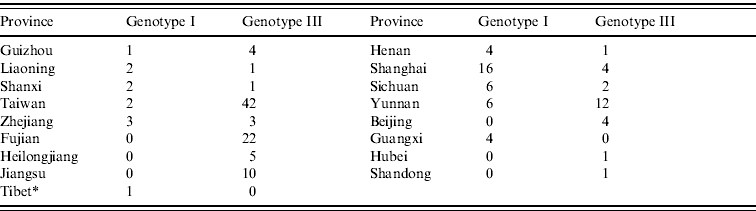
* Genotype V (strain XZ0934) was also circulated in Tibet.
Selection pressure
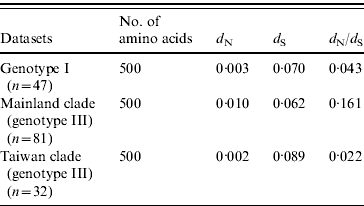
The selection pressure acting on the envelope gene of JEV as measured by average d N/d S values are shown in Table 2. There were no significantly synonymous variations in the three datasets. However, the non-synonymous variation of mainland clade was much higher, characterized by the highest d N/d S value, being 7·3 times higher than Taiwan clade and 3·7 times higher than ‘genotype I’. These data suggest that mainland clade was under weaker purifying selection compared to genotype I and the Taiwan clade.
Table 2. Selection pressure acting on the envelope gene
Site-specific selection pressure analysis (Table 3) was also performed by using the Datamonkey web server. The number of codons that exhibited patterns of positive selection was five (aa 6, 166, 312, 433, 498) and three (aa 132, 143, 306) for the genotype I dataset and the Taiwan clade datasets, respectively, using one detection method (REL). However, in mainland clade, two codons (aa 227, 408) were identified as being under strong positive selection using two detection methods (FEL, IFEL), and one codon (site 227) was identified using three detection methods (SLAC, FEL, IFEL).
Table 3. Positive selection sites analysis by using SLAC, FEL, IFEL, and REL methods

SLAC, Single-likelihood ancestor counting; FEL, fixed-effects likelihood; IFEL internal fixed-effects likelihood; REL, random-effects likelihood.
Bold values denote amino-acid sites under strong positive selection.
* Amino acid site where P<0·1.
† Amino acid site where Bayes factor >50.
‡ n.a., Not analysed due to alignment size restriction.
Rates of evolutionary change
As shown in Table 4, our MCMC analysis revealed that rates of nucleotide substitution in the Taiwan clade and genotype I were 8·4 times and 8·9 times higher, respectively, than those in the mainland clade.
Table 4. Evolutionary processes on envelope protein of JEV
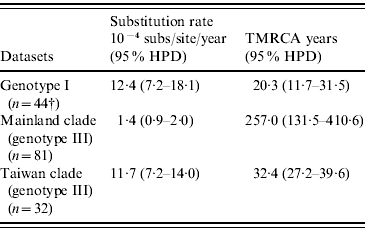
TMRCA, Time to the most recent common ancestor; HPD, high probability density.
† Among genotype I, only three strains were isolated before 1987 and all other 44 strains were isolated after 2000. Because of serious sampling bias, evolutionary analysis was performed by excluding three strains isolated before 1987.
Times of common ancestry
Table 4 also showed TMRCA years of genotype I, mainland clade, and Taiwan clade. As for genotype I and Taiwan clade, they were newly introduced in 1989 (age 20·3 years, 95% HPD 11·7–31·5) and 1976 (age 32·4 years, 95% HPD 27·2–39·6), respectively. However, TMRCA of mainland clade was aged 257·0 years (95% HPD 131·5–410·6).
DISCUSSION
Japanese encephalitis (JE) is an important mosquito-borne disease in China. Although JEV-infected cases decreased markedly after the nationwide vaccination programme initiated in the 1970s, reported cases are still very high, with 7860, 5422, 5097, 7643, 4660, 2975, 3913, and 2738 cases from 2003 to 2010 (www.chinacdc.cn), respectively. According to the case report from 1998 to 2002, provinces are classified into high-prevalence (JE incidence >1/100 000), medium-prevalence (0·5–1/100 000), and low-prevalence (<0·5/100 000) areas [Reference Gao20]. Of those provinces mentioned in this study, Shanxi, Henan, Sichuan, Guizhou, and Yunnan belong to high-prevalence areas; Hubei and Guangxi belong to medium-prevalence areas; and Shandong, Jiangsu, Zhejiang, and Fujian belong to low-prevalence areas. According to data presented in this study (Fig. 2), JEV strains are co-circulating not only in high-prevalence areas (Shanxi, Henan, Sichuan, Guizhou, Yunnan), but also in low-prevalence areas (Zhejiang). Specifically, two genotypes were even simultaneously isolated in Yunnan (1979, 1986), Zhejiang (2008), Taiwan (2008), and Tibet (2009), as shown in Figure 1. This suggests that there are probably no predominant genotypes in China.
Although genotypes I and III are co-circulating in China, genotype I and the Taiwan clade of genotype III were newly introduced around 1989 and 1976, respectively (Table 4). Regarding genotype I, it spread widely over 20 years, from south China (Yunnan) to north China (Liaoning) and from east China (Shanghai, Taiwan) to central China (Sichuan). However, this genotype (TPC0806c and YL0806f) appeared in Taiwan only in 2008 (Fig. 1). They were probably imported from adjacent provinces like Shanghai and Zhejiang by water birds [Reference Huang10]. The late introduction might be due to low accommodation of genotype I in the local environment. Similarly, newly introduced Taiwan clade does not circulate in the mainland, which might be due to low accommodation of the virus in the local environment. Therefore, it appears that some factors prefer different cell tropism, low replication rate in the hosts, different mosquito populations, or adverse conditions associated with winter, dry season, etc. might explain the low ability of some JEV strains to survive after importation.
Conversely, strains in the mainland clade have simultaneously circulated in the mainland and Taiwan since at least 1972 (Fig. 1). The Taiwan strains in the mainland clade and some strains isolated in adjacent provinces (Fujian, Zhejiang, Jiangsu, Shanghai) fall into the same subgroup. Just like genotype I, these Taiwan strains might also have been introduced from the mainland. Therefore, the mainland clade and Taiwan clades are helpful in elucidating how the virus adapts to the environment after importation based on their different accommodation abilities.
The evolutionary rates shown in Table 4 all fall within the range seen in other RNA viruses [Reference Jenkins26]. However, the evolutionary rates of the newly introduced Taiwan clade and genotype I are much higher than the mean evolutionary rates of genotypes III and I in Asia [Reference Pan27]. Specifically, the Taiwan clade and genotype I evolve 8·4 times and 8·9 times faster than the mainland clade. Faster evolutionary rates could be related to a faster replication cycle of the virus, particularly tropism, and transmission modes [Reference Hanada28, Reference Gray29]. Therefore, faster evolutionary rates of the Taiwan clade and genotype I could be a prerequisite for these strains to inhabit a new environment after importation.
Another finding in this study is the positive selection in the JEV envelope protein. In a previous study [Reference Yang30, Reference Tang31], JEV was shown to be under strong purifying selection but no positive selection sites were identified. The only confirmed positive selection site is located within NS4B (aa 24) [Reference Schuh32]. However, we demonstrate in this study that JEV strains in China are not only under purifying selection, but also probably under positive selection (Tables 2 and 3). Specifically, there are evidences of positive selection acting on strains in the mainland clade, especially aa 227 and 408 in the envelope protein. As for the Taiwan clade and genotype I, there are three and five potential positive selection sites, respectively. This shows that mutations in the JEV envelope protein tend to be beneficial rather than neutral or deleterious, although the local strains are under strong purifying selection. Since the JEV envelope protein is involved in virus entry and assembly [Reference Rey33], the positive selection might be of helpful for the virus to replicate efficiently in hosts (human, mosquitoes, birds, pigs, etc.).
In conclusion, we have elucidated the molecular epidemiology and evolution of JEV in China. Specifically, genotypes I and III are co-circulating not only in high-prevalence areas, but also in low-prevalence areas. Compared to the mainland clade of genotype III, genotype I and the Taiwan clade of genotype III are newly introduced and evolving more rapidly. Meanwhile, local JEV strains, especially those in the mainland clade, are confirmed as likely to be under positive selection.
ACKNOWLEDGEMENTS
The author thanks Liqun Fang for drawing the maps.
DECLARATION OF INTEREST
None.






