Introduction
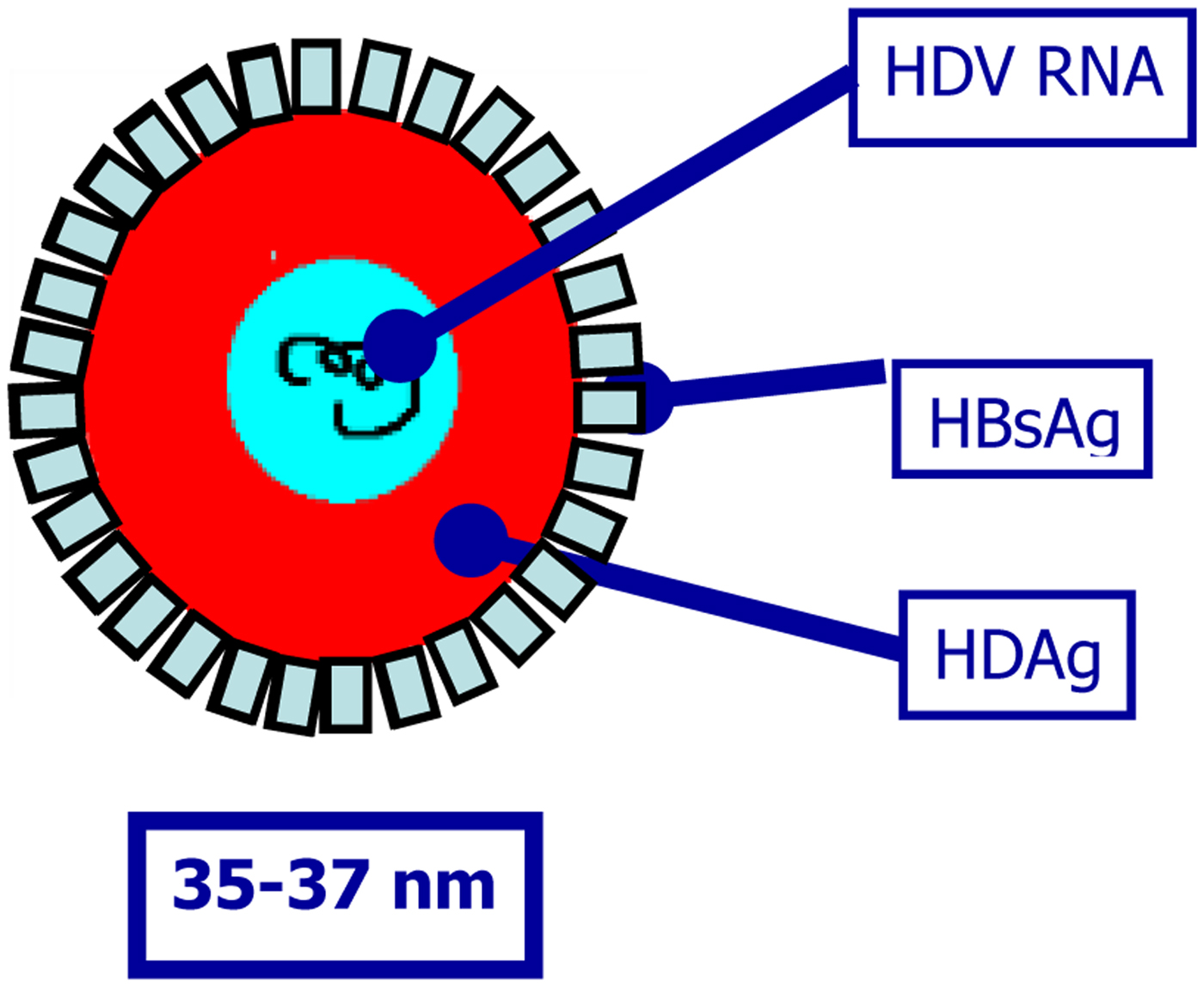
Hepatitis delta virus (HDV) is a defective RNA virus that requires the presence of hepatitis B virus (HBV) for virion assembly and propagation [Reference Sureau, Guerra and Lanford1–Reference Taylor, Fields, Knipe and Howley5]. It is a small RNA virus with a circular RNA genome of approximately 1.7 Kb in length (Fig. 1) [Reference Flores, Ruiz-Ruiz and Serra6]. The virus does not code any enzyme to replicate its genome using instead RNA polymerase II from hepatocytes for the synthesis of its RNAs, both with positive and negative polarity. HDV encodes a single ORF responsible for the expression of the delta antigen (HDAg), in two isoforms, the small (S-HDAg) and large HDAg (L-HDAg) [Reference Shirvani-Dastgerdi and Tacke7]. HDAg and HDV RNA are subsequently encased in an envelope embedded with HBV surface antigen (HBsAg) to form infectious virions (Fig. 1) [Reference Shirvani-Dastgerdi and Tacke7]. As a consequence, the early step of HDV entry into hepatocytes follows the same mechanism for both viruses. Once in the hepatocytes, a nuclear localisation signal on L-HDAg triggers the translocation of HDV nucleocapsid to the nucleus where viral genome is replicated.

Fig. 1. Hepatitis delta virus virion.
HDV infection can be acquired either as coinfection of the two viruses or as superinfection of an already chronic carrier of HBV. Normally, in coinfection, HDV activation depends on HBV activation. As a consequence, HDV expression in coinfection can vary widely, ranging from an abortive infection to an extremely virulent form of disease [Reference Casey8–Reference Stroffolini10]. In the vast majority of coinfections, however, HBV infection is self-limiting and HDV infection recovers, with a progression to a chronic disease in about 2% of cases, only [Reference Caredda11].
Superinfection with HDV in chronic hepatitis B causes a more threatening form of liver disease that increases the liver damage leading to more rapid progression to cirrhosis in up to 90% of cases [Reference Smedile12]. Early studies in the general population demonstrated a benign, non-progressive disease in about 15% of cases, while the vast majority of cases experience a rapid progression to cirrhosis within few years [Reference Rizzetto13]. Subsequent studies demonstrated that HDV-positive cirrhotic patients had significantly higher risk of progression towards hepatocellular carcinoma (HCC) development and liver decompensation.
This article provides an update on the relationship between HDV infection and development of HCC, highlighting the possible mechanisms of carcinogenesis. We also review the most recent epidemiological data as well as advances in HDV treatment.
Epidemiology of HDV-related HCC
Liver cancer is an important health problem worldwide, being the second cause of cancer-related death. More than 90% of the primary liver cancers are HCC having a particularly poor prognosis, with 700 000 new cases and 600 000 deaths occurring every year [Reference Petruzziello14]. Infections with hepatitis B, C and delta viruses, all having a great propensity to progress towards chronicity, are the aetiological agent of more than 80% of all HCCs. It has been calculated that approximately 170 million chronic HCV carriers, 350 million chronic HBV carriers and 15–20 million HDV carriers represent the burden of chronic hepatitis worldwide [Reference Petruzziello14, Reference Petruzziello15]. HBV/HDV coinfected patients experience rapid progression to more advanced stages of liver disease and also have the highest mortality rate (20%) of any of the viral hepatitis [Reference Bray16–Reference Farci and Niro18].
Several mechanisms for HCC development and progression in the presence of viral hepatitis have been proposed. These include antiviral inflammatory response, immune clearance of infected cells and subsequent hepatocytes regeneration all leading to genetic and epigenetic changes, predisposing to HCC development [Reference Buti19–Reference Mesri, Feitelson and Munger22].
The International Agency for Research on Cancer (IARC) has recently categorised several infectious agents into four groups, according to the evidence for carcinogenicity to humans [23]: group 1 (high evidence of carcinogenicity to humans), group 2A (possibility of carcinogenic effects), group 2B (low carcinogenic effects) and group 3 (not sufficient evidences of carcinogenicity). While HBV and HCV are both allocated in IARC group 1, HDV was allocated several years ago in group 3 [24], as the demonstration of HDV contribution to HCC induction on HBV was not adequate. However, more recently, it has been suggested that the risk of HCC is higher when HBV is superinfected by HDV [Reference Ghamari25].
The analysis of several epidemiological studies published lately has shown the existence of some controversies about the increasing risk of HCC development in HDV chronically infected patients. An important study by the European Concerted Action on Viral Hepatitis (Eurohep) demonstrated that HBV/HDV-positive cirrhotics followed for a median 6.6 years had a twofold mortality risk than HBV cirrhosis. Moreover, the estimated risk for HCC was 13% for HBV/HDV cirrhotics as compared with 2–4% for HBV cirrhotics, thus increasing the risk to threefold [Reference Fattovich26]. A retrospective study on data collected in South London showed that despite the increasing prevalence of HDV infection, the risk of HCC was similar in HDV-positive and negative patients [Reference Cross27]. Very high prevalence (18.8% and 23%) of HDV markers in HCC patients was shown in two Turkish studies [Reference Uzunalimoğlu28, Reference Değertekin, Yalçin and Yakut29], as well as in a small study published in Jordan [Reference Toukan30].
We published the results of 299 patients with chronic HDV infection followed for a mean interval of 28 years. The study demonstrated that 30% of patients developed cirrhosis and <15% of all patients died of liver-related causes [Reference Romeo31]. These results are in contrast to the data published previously but more in line with subsequently published results, suggesting that our cohort probably reflected a change in the epidemiology of the infection, but also one could suggest that our patients were rather the result of a long-lasting asymptomatic disease that eventually lead to cirrhosis than a recent rapidly progressive infection. This same conclusion had already been suggested by others indicating a biphasic course of HDV-related disease, characterised by an initially florid infection and a rapid progression towards liver failure with high mortality rates [Reference Rosina32]. Patients surviving the initial phase experienced a slowly progressing disease leading to liver decompensation later [Reference Ji, Sundquist and Sundquist33]. Our results also demonstrated that persistent HBV/HDV infection was associated with cirrhosis development and HCC occurrence at 4% and 2.8%, respectively. Moreover, persistent HDV replication was the only predictor of liver-related mortality [Reference Romeo31].
Similar results as ours were obtained by Buti et al. [Reference Buti19] in a cohort of 158 chronic HDV patients, followed for a median of 158 months. In this study, 18% experienced liver decompensation, while 3% developed HCC. A recently published study calculated the standardised risk (standardised incidence ratios (SIRs)) for HDV patients of developing HCC, demonstrating an increased risk for HDV as compared with HBV (SIR 6.11, 95% CI 2.77–11.65) [Reference Ji, Sundquist and Sundquist33].
It has been recently demonstrated that high levels of HDV replication in non-cirrhotic patients are associated with an increasing progression to cirrhosis and to HCC development (multivariate analysis OR 1.42, 95% CI 1.04–1.95; P = 0.03), while the role of HDV viraemia as a predictor of negative outcome in cirrhotic patients lessens [Reference Romeo34].
In a study from Japan, HDV superinfection increases the risk of cirrhosis and HCC. The proportion of HCC per 1000 person years was 7.84 among cases with anti-HDV and 2.73 among those without anti-HDV. The overall relative risk of HCC was 2.87, 95% CI 1.03–6.23 [Reference Tamura35]. A study from Taiwan failed to show any acceleration in the development of HCC in patients with HDV superinfection. Nevertheless, the numbers of patients in HDV group were small compared with HBV monoinfection group (42 vs. 255) [Reference Huo36].
A recently published case–control study aimed at the determination of the role of HBV, HCV and HDV in HCC development in Cameroon, Central Africa [Reference Huo36]. The sera of 88 consecutive HCC patients and 85 controls without known liver disease were analysed for the presence of markers of viral hepatitis. The analysis of risk factors associated with the development of HCC demonstrated a strong association with the presence of HBV, HCV and HDV markers, being the OD (95% CI) of 16.3 (7.2–37.1), 9.6 (2.8–33.6) and 29.3 (4.1–1231) for the three viruses, respectively.
Finally, a very recent publication on the epidemiological characteristics of HDV infection on a nationwide Swiss HIV Cohort Study, aiming at the evaluation of the impact and clinical outcome of the infection, indicates a prevalence of HDV infection of 15.4% (119/771, 95% CI 12.9–18.0) in HIV-infected patients, with HDV replication in 62.9% [Reference Amougou37]. Moreover, HDV infection appeared as strongly associated with overall death (adjusted hazard ratio 2.33, 95% CI 1.41–3.84), liver-related death (7.71, 3.13–18.97) and HCC occurrence (9.30, 3.03–28.61). Noteworthy, results were similar when HCV-coinfected or people injecting drugs were excluded from the analysis.
Taken together, the results of the above-mentioned studies, summarised in Table 1, emphasise the need for developing new treatments for chronic HDV infection as the presently available drugs are largely ineffective for viral eradication and control of disease progression.
Table 1. The epidemiological studies on the role of hepatitis D virus infection in increasing the risk of hepatocellular carcinoma

Treatment of HDV infection
Ideally, a successful treatment for chronic HDV infection would eradicate HDV as well as its helper virus. Clearance of HDV is considered as obtained when both HDV RNA and liver HDAg become persistently undetectable. A complete resolution is achieved when HBsAg clearance is obtained [Reference Béguelin38]. The persistence of HBsAg corresponds to an HDV-persistent infectivity, even with low viral titres. Unfortunately, despite inhibition of HBV replication and, in few cases, HBsAg clearance, no clearance of HDV has been observed when oral antivirals against HBV were used alone. Lamivudine, ribavirin, famciclovir, adefovir and entecavir have shown little or no effect when used alone in HDV treatment, regardless of the length of drug administration [Reference Alves, Branco and Cunha39–Reference Lau43].
Interferon (IFN) is still the only recognised treatment for HDV chronic infection, even though results published over the years have demonstrated that this drug is far from being the optimal treatment [Reference Wedemeyer44]. Response to treatment is extremely variable and it may occur even spontaneously, at the end of treatment, in analogy with what already known for HBV infection. The response is normally proportionate to drug dosage, as demonstrated by an early publication, demonstrating higher efficacy of 9 MU of INF administered subcutaneously thrice a week, as compared with 3 MU thrice a week [Reference Niro, Rosina and Rizzetto45]. The results of this study demonstrated that by the end of treatment, negative HDV RNA and normal ALT were found in 71% and 71%, respectively, of patients treated with higher dosage, against 36% and 29%, respectively, obtained in patients treated with lower IFN dose [Reference Lau43].
By 2006, IFN-α was largely replaced by pegylated IFN (PEG-IFN) [Reference Wedemeyer44–Reference Farci46] with improved pharmacokinetic. Studies of PEG-IFN in hepatitis delta have shown HDV RNA suppression during treatment in 17–47% of patients, with a post-treatment week 24 virological response, observed in 25–40% of patients [Reference Béguelin38, Reference Yurdaydin42, Reference Wedemeyer44]. Several studies have emphasised the importance of negative HDV RNA after 6 months of treatment as a predictor of response at 12 months, selecting patients that should be treated for longer periods [Reference Yurdaydin42, Reference Lau43, Reference Farci46]. The analysis of factors as potential predictors of response has indicated that naïve patients with low levels of serum GGT appeared to have a better response [Reference Niro47]. More recently, several publications have demonstrated that the therapeutic effect of PEG-IFN combined to several analogues does not increase the proportion of patients with complete HDV eradication [Reference Garripoli41, Reference Wedemeyer44, Reference Castelnau48–Reference Yurdaydin51]. One possible explanation could be that since these drugs are not effective as inhibitors of the only HBV component necessary for HDV survival, HBsAg, HBV DNA suppression alone does not lead to HDV eradication.
However, recently published findings have pointed out that in spite of the disappointing results so far shown, IFN treatment is independently associated with a more benign clinical long-term outcome [Reference Wedemeyer, Yurdaydin and Caruntu52]. Indeed, the results of this retrospective single-centre study demonstrate that the frequency of hepatic decompensation is reduced in IFN-α treated patients, while there is no difference in terms of HCC development. Finally, loss of HDV RNA by IFN-α did not influence HCC development [Reference Wranke53].
Recently, new strategies for HDV treatment are under investigation. Being HDV infection always associated with HBV presence, it must be considered that the two viruses may interfere with each other at several steps of the replication cycles. The optimal treatment should have as a gold standard the loss of HBsAg, resulting in an automatic eradication of HDV replication and propagation. With this in mind, the new therapeutic approaches act at different levels of HDV lifecycle. The sodium taurocholate co-transporting polypeptide (NCTP) receptor and the myristoylated N-terminal pre-S1 domain of the L-HBsAg are essential for viral entry into cells [Reference Meier54, Reference Zhong55]. Myrcludex B is a mirystoylated lipopeptide that has been demonstrated as capable of blocking HBV entry into hepatocytes [Reference Ni56]. In vitro studies have shown that myrcludex B is able to inhibit HDV entry as well [Reference Li and Urban57]. Interim results of a phase II study on the administration of myrcludex B in humans have been recently published [Reference Schulze58]. This study indicates that myrcludex B is associated with HDV RNA decline in a good proportion of patients but the result is encumbered HDV RNA reappearance after drug stop, in a good proportion of cases. Once the virus is within the hepatocyte, the replicative cycle happens through the host polymerase, giving rise to L-HDAg that will later interact with HBsAg, forming new viral particles. Prenylation of the newly formed L-HDAg is required for particle formation. The inhibition of such a step prevents the appearance of new viral particles [Reference Bogomolov59, Reference Bordier60].
Finally, once the new viral particles are formed, they are released from the cell. At this stage, very recent studies indicate that the use of nucleic acid polymers (NAPs) can block HBsAg release by binding to class I surface glycoproteins [Reference Koh61]. Few studies have already been published on the administration of NAPs, demonstrating serum HBsAg and HBV DNA reduction with viral rebound occurring in the majority of cases after treatment stop [Reference Kocisko62].
Overall, the treatment results so far presented indicate that we are still not ready for a curative treatment against HDV infection, leaving the road open to progression towards more severe clinical outcomes, with HCC development as the final step.
Oncogenic mechanisms
It is still not clear whether HDV adds additional oncogenic effects beyond promoting fibrosis deposition and development of cirrhosis. HDAg expression alone does not appear to be cytopathic and does not appear to have oncogenic potential. On the other hand, it has been indicated that since high levels of antigen and viral RNA cause cell cycle arrest in G1 phase, this mechanism could be responsible for cell death in the acute phase of the infection, when viral replication is high [Reference Taylor4]. However, the intrahepatic expression of L-HDAg appears to be associated with high levels of liver inflammation through NF-κB activation signalling and the immune response against the virus [Reference Al-Mahtab, Bazinet and Vaillant63, Reference Guilhot64]. These proteins seem to be implicated in cell transformation and tumourigenesis, indeed STAT3 over expression is associated with leukaemia, prostate cancer and melanoma [Reference Negro65, Reference Benekli66]. Niu et al. [Reference Niu67] demonstrated that L-HDAg (p27) significantly increases the NFκB activity also via tumour necrosis factor α (TNFα), TNF receptor-associated factor (TRAF2), IKKβ and p65-mediated induction. Park et al. [Reference Park68] have shown that HDV p27 activates STAT3 via phosphorylation of tyrosine 705 residue. STAT3, in turn, regulates DNA methyltransferases (DNMT1) and causes the overexpression of DNMT3b. Since DNMT1 is responsible for the maintenance of methylation patterns, whereas DNMT 3a and 3b catalyse new methylation events, their overexpression can be potentially oncogenic.
Interestingly, it has been postulated an epigenetic control of HDV over HBV transcription and regulation, that would explain why HBV replication is inhibited in the presence of HBV, implying an HCC development as depending on HDV alone [Reference Benegiamo69]. Against this hypothesis is the evidence that long-lasting active proliferation of the two viruses leads to more aggressive disease and HCC development as already mentioned.
Recently, it has been suggested that long non-coding RNAs (lncRNAs) may play a role in the pathogenesis of HDV-related HCC, through the dysregulation of lncRNAs unique to HDV [Reference Dastgerdi, Herbers and Tacke70].
Early studies demonstrated that the lymphocytes-mediated cell lysis has a non-relevant role in determining liver disease, indicating that HDV is not able to raise an autoimmune response [Reference Zhang71]. By the same token, since lymphocytes T CD4+ specific for HD antigen have been observed within the liver of chronically HDV-infected patients, it has been postulated that T lymphocytes-specific clones against HD antigen may be involved in B and T cellular response, leading to the immune activation against HDV [Reference McFarlane72]. Finally, more recent findings again propose that hepatitis D is an immune-mediated disease, showing a CD4+ T cell rise in HDV infection [Reference Nisini73]. Since several autoantibodies are commonly detected in HDV-infected patients, it is still not completely understood whether this represents an accompanying pattern rather than a true autoimmune manifestation. As long as we do not have conclusive evidences for or against the true pathogenic mechanism of HDV-related liver disease, we keep exploring several different mechanisms as possibly involved in HCC development.
In conclusion, the mechanism by which HDV causes HCC still remains to be elucidated, even if several events occurring during HDV infection, like oxidative stress as result of the severe necroinflammation HDV-related, the aberrant silencing of tumour suppressor genes by DNA methyltransferases, increased levels of histone H3 acetylation of the clusterin promoter and the targeted inhibition of STAT3 and cyclophilin might have a potential role in explaining the oncogenetic role of HDV [Reference Wedemeyer and Manns74].
Genetic heterogeneity
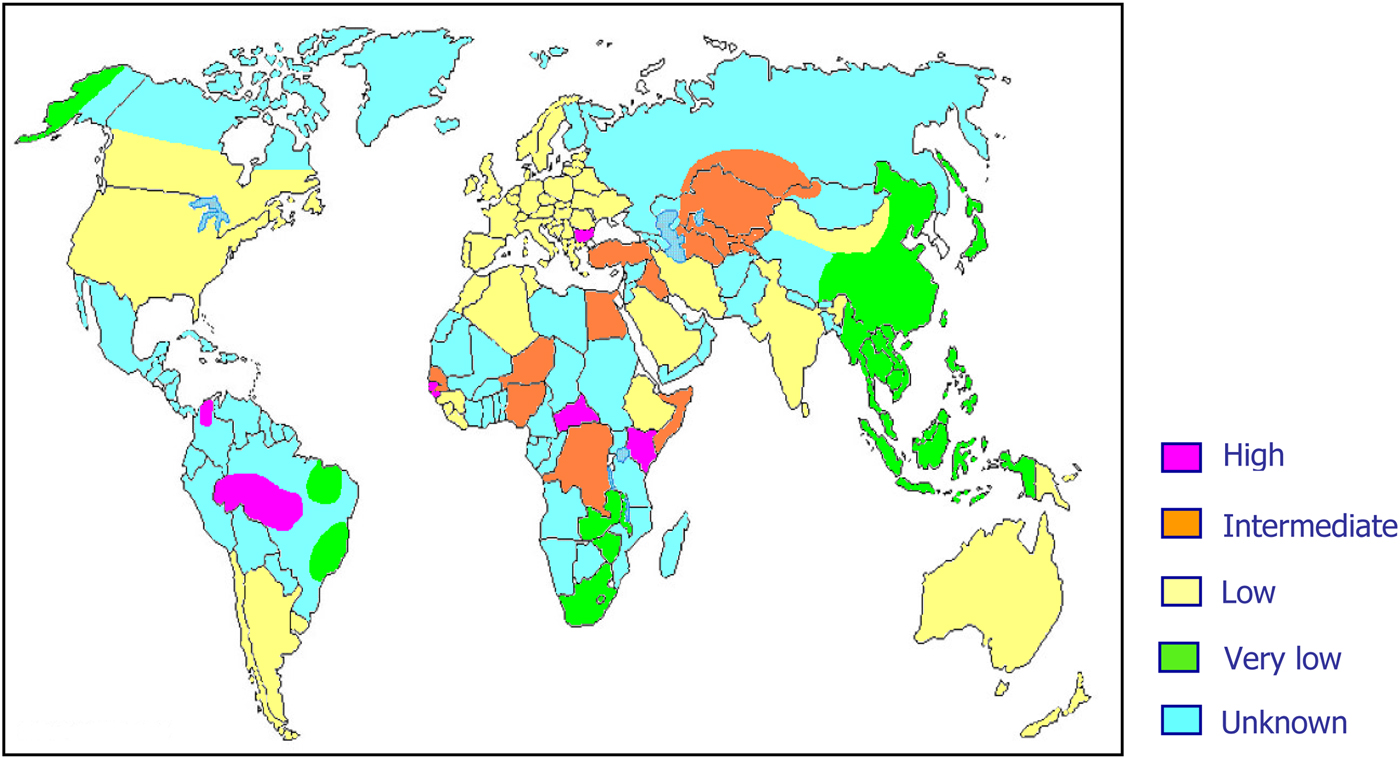
As previously mentioned, about 15–20 million people worldwide are considered to be chronic HDV carriers. However, it is highly probable that these data are largely underestimated mainly because of lack of data on HDV prevalence from several areas of the world (Fig. 2).
Fig. 2. World prevalence of hepatitis delta virus infection.
HDV RNA, like all RNAs, is characterised by high degree of genetic variability, resulting in at least eight major clades distributed worldwide [Reference Abbas75].
The pathogenetic role of viral heterogeneity in HDV-related clinical outcome is still unknown. However, early data have demonstrated that HDV genotype III, endemic in the Amazonian area and northern South America, is associated with very aggressive forms of liver disease [Reference Casey8, Reference Radjef76], while genotype II, prevalent in Far East and Japan, is associated with a milder clinical course [Reference Nakano77] and genotype I, present mostly in Europe and North America, is associated with a wide variety of clinical conditions [Reference Lee78].
A recent finding has demonstrated the occurrence of a natural intergenotypic recombination between HDV genotypes I and II [Reference Niro79, Reference Sy80], suggesting that the occurrence of mixed genotypes as well as recombinant HDV genotypes may be responsible for alterations leading to different clinical outcomes and possibly HCC development.
Conclusions
HDV remains a potential risk factor for serious liver damage, either for HBV chronic carriers or for all those still susceptible to HBV infection, for whom HBV vaccination is strongly recommended. Early long-term treatment of chronic HDV infection with IFN still offers better chances of infection control, lowering the risk of progression to cirrhosis and liver cancer. A better understanding of the mechanisms responsible for HCC development in chronic HDV infection is necessary also for directing research toward the development of efficient antiviral compounds able to eradicate the infection and redesign the natural history of the disease. A global effort is deemed urgent to enhance the models already existing as well as to learn more about viral infection and correlated tumorigenesis mechanisms.
Author ORCIDs
Raffaella Romeo (0000-0002-7858-4670); Arnolfo Petruzziello (0000-0003-0353-9151); Eve Isabel Pecheur (0000-0002-8613-862X); Floriana Facchetti (0000-0001-5252-4744); Riccardo Perbellini (0000-0002-4412-1256); Enrico Galmozzi (0000-0001-8831-2401); Najeeb Ullah Khan (0000-0003-2568-9484); Lucia Di Capua (0000-0002-6113-688X); Rocco Sabatino (0000-0003-1113-8665); Gerardo Botti (0000-0002-6287-733X); Giovanna Loquercio (0000-0002-7539-2075).
Author contributions
Romeo R, Facchetti F, Galmozzi E, Perbellini R, Di Capua L and Sabatino R acquired the data; Romeo R drafted the article and contributed to the conception and design in collaboration with Loquercio G; Botti G, Petruzziello A and Pecheur EI contributed to the critical revision for important intellectual content; all authors approved the final version to be published.
Conflict of interest
None.
Data sharing statement
Participants gave informed consent for data sharing.