


Integrins, the major cellular receptors for proteins of the extracellular matrix (ECM), are heterodimers consisting of one α and one β subunit. In mammals, eight β subunits and 18 α subunits combine to create 24 different heterodimers. Both the α and β subunits contribute to ligand-binding specificity (Refs Reference Humphries, Byron and Humphries1, Reference Hynes2). While some integrin subunits pair with only one partner, others are promiscuous. For example, the β1 (ITGB1) subunit pairs with the α1–α11 (ITGA1–11) subunits, as well as αv (ITGAV). The resulting β1 integrins can be grouped by ligand-binding specificity. Integrins α3β1, α6β1 and α7β1 bind laminin isoforms; integrins α5β1, α8β1 and αvβ1 bind the Arg-Gly-Asp (RGD) motif found in fibronectin (FN1) and other matrix proteins; integrins α4β1 and α9β1 bind the Leu-Asp-Val (LDV) motif also present in fibronectin and other matrix proteins; and integrins α1β1, α2β1, α10β1 and α11β1 bind collagens (Refs Reference Humphries, Byron and Humphries1, Reference Hynes2). Several of the collagen-binding integrins can also recognise binding sites in certain laminin isoforms, but for the purpose of this review, laminin-binding integrins refers to the subfamily containing the related α3, α6 and α7 subunits.
The data summarised above paint a complex picture in which many integrins recognise more than one ligand and many ligands contain binding sites for more than one integrin. In addition, they point to a conundrum regarding the molecular basis of functional diversity within specific classes of integrins. Upon ligand binding, different integrins transduce signals that differently influence cell survival, proliferation, cytoskeletal organisation, cell adhesion, and migration. Although a few cytoplasmic effectors have been identified that interact with integrin α subunits, the substantial majority associate directly or indirectly with the cytoplasmic tails of integrin β subunits (Refs Reference Liu, Calderwood and Ginsberg3, Reference Miranti and Brugge4). Thus, a question that arises for β1 integrins is how so many different heterodimers, each with a common β1 signalling subunit, are able to mediate diverse cellular responses to different ECM proteins.
Here I review how the cell behaviours and signalling pathways mediated by the α3/α6/α7 laminin-binding integrin subfamily differ from those mediated by other integrins, and discuss how these functional and cell biological differences may relate to the unusual ability of the laminin-binding integrins to interact with members of the tetraspanin family of proteins. The α6 subunit is further distinguished by its ability to pair with the β4 (ITGB4) subunit. The unique properties of α6β4 integrin, which associates with tetraspanins as well, are also discussed in the review. Tetraspanin association has significant implications not only for the complex roles the laminin-binding integrins play in metastasis but also for how these integrins might ultimately be targeted in anticancer therapies.
Distinct cell responses mediated by laminin-binding integrins
Early recognition that cellular responses mediated by laminin-binding integrins might be different from those mediated by other integrins came from studies of neurons. Dorsal root ganglion and olfactory epithelial neurons both displayed preferential migration on laminin isoforms compared with other ECM proteins, such as collagens and fibronectin (Refs Reference Gundersen5, Reference Calof and Lander6). Moreover, as observed by interference reflectance microscopy, neuronal cells made fewer and smaller close contacts with laminin substrates than they did with fibronectin substrates (Refs Reference Calof and Lander6, Reference Gundersen7). The organisation of focal adhesions – the cytoskeletal and signalling complexes that form at sites of integrin adhesion to ECM – also appeared different on laminin: neurons formed prominent focal adhesions on fibronectin, but on laminin they formed smaller and less numerous focal-adhesion-like structures (Ref. Reference Gomez, Roche and Letourneau8).
More recently, results similar to those described for neuronal cells have been obtained for tumour and epithelial cells. Lung adenocarcinoma cells, squamous carcinoma cells, and nontransformed keratinocytes all migrated up to fivefold faster on laminin isoforms than on other ECM ligands such as fibronectin or collagen I (COL1A1) (Refs Reference Gu9, Reference Frank and Carter10, Reference Zhou and Kramer11, Reference Winterwood12). On fibronectin or collagen I, cells formed large actin stress fibres and prominent focal adhesions, while on laminins fewer stress fibres and smaller, more-peripheral focal-adhesion-like structures (sometimes termed ‘focal contacts’) were observed (Refs Reference Gu9, Reference Frank and Carter10, Reference Zhou and Kramer11). Thus, data from both neuronal and tumour cell systems converge on the view that laminin-binding integrins frequently mediate distinct cell behaviours typified by rapid migration, smaller focal complexes, and fewer actin stress fibres.
In principle, the functional differences between laminin-binding integrins and other integrins could result from differences in ligand-binding affinities. However, the dissociation constants (K ds) of activated laminin-binding integrins for different laminin isoforms are in the range of ~1–20 nm (Ref. Reference Nishiuchi13), which is similar to the K d of activated α5β1 for fibronectin (~10 nm) (Ref. Reference Faull14), and the Kds of activated α1β1 and α2β1 for collagen ligands (~1–10 nm) (Ref. Reference Kern15). Instead, it appears that laminin-binding integrins may signal differently than other integrins. In lung adenocarcinoma cells, adhesion on fibronectin triggered strong activation of the RhoA (RHOA) small GTPase, with little activation of the Rac1 (RAC1) small GTPase (Ref. Reference Gu9). Conversely α3β1-integrin-dependent adhesion on laminin-10/11 (termed laminin-511/521 when named according to its composition of α, β and γ chains) triggered Rac activation with very little concomitant RhoA activation. In addition, focal adhesion kinase (FAK/PTK2) activation was significantly higher in cells on fibronectin than in cells on laminin-10/11 or laminin-5 (laminin-332), while formation of the trimolecular complex of p130CAS (BCAR1), CrkII (CRK) and DOCK180 (DOCK1), which promotes Rac1 activation and cell migration, was much higher on laminin isoforms than on fibronectin (Ref. Reference Gu9). Similarly, α3β1-integrin-dependent adhesion of squamous carcinoma cells on laminin-5 resulted in less RhoA activation and enhanced activation of the small GTPase Cdc42 (CDC42) and its effector PAK1, compared with cells adherent on collagen I (Ref. Reference Zhou and Kramer11).
Collectively, these observations suggest that, compared with other integrin ligands, cellular responses to laminin isoforms involve a different profile of signalling through small GTPases in the Rho family, which control the organisation of the actin cytoskeleton. Signalling through laminin-binding integrins often features minimal activation of RhoA, which controls the formation of actin stress fibres and prominent focal adhesions, and strong activation of Rac1 and Cdc42, which control the formation of lamellipodia and filopodia, respectively. The result is smaller, less numerous focal contacts, and a dynamic actin cytoskeleton oriented towards rapid migration rather than strong substrate adhesion.
With the strikingly rapid migration that laminin isoforms such as laminin-5 can provoke in vitro, an attractive hypothesis is that laminin-binding integrins are poised to be potent mediators of tumour cell motility, migration and invasion during metastasis. Indeed, several studies support the hypothesis that laminin-binding integrins can have prometastatic functions. However, the roles of laminin-binding integrins in metastasis are more complex than such a straightforward hypothesis would suggest, and, depending on the context, the laminin-binding integrins may in fact possess antimetastatic functions. In the following sections, potential pro- and antimetastatic functions of the laminin-binding integrins are discussed, with an emphasis on in vivo experimental results and clinical correlations.
Potential roles of α3β1 integrin in metastasis
Although several studies have identified a positive correlation between tumour cell expression of α3β1 integrin and propensity for metastasis, a roughly equal number of studies have reported negative correlations (Table 1). Moreover, in some cases, such as breast carcinoma and oral squamous cell carcinomas, both positive and negative correlations have been reported. These conflicting data suggest that the role of α3β1 integrin in metastasis may be highly dependent on context, even within the same general class of tumours.
Table 1. Clinical or in vivo experimental studies reporting correlations between α3 integrin expression and metastasis

Underlying the potential prometastatic activities of α3β1 integrin might be its ability to promote adhesion, migration and survival in response to its ligands laminin-5 and laminin-10/11, which are present in basement membranes beneath epithelial or endothelial cell layers. Indeed, anti-α3-integrin function-blocking antibodies have inhibited metastatic cell behaviours in several in vivo models, including brain colonisation by non-small-cell lung carcinoma cells inoculated intracardially (Ref. Reference Yoshimasu16), colonisation by gastric carcinoma after intraperitoneal implantation (Ref. Reference Takatsuki17), lung colonisation by fibrosarcoma cells upon tail-vein injection (Ref. Reference Wang18), and invasion of glioma cells after intracerebral implantation (Ref. Reference Kawataki19). The design of these experiments, in which cells were treated with antibody once, just prior to implantation, suggests that acutely blocking α3β1 function can in some cases inhibit the ability of circulating tumour cells to adhere, invade, survive or proliferate at a secondary site.
How α3β1 may act in other settings to suppress metastasis is not well understood. Although not typified by prominent focal contacts, cell–substrate adhesion mediated by α3β1 could still in some cases prevent tumour cells from detaching from the primary tumour site. Selection of PC-3 prostate carcinoma cells with enhanced invasive potential yielded a subpopulation with low expression of α3 integrin, suggesting that reduced α3-dependent adhesion could promote enhanced mobility in some settings (Ref. Reference Dedhar20). Analysis of renal carcinoma cells shed from primary tumours revealed reduced α3 integrin expression on the circulating tumour cells, suggesting a role for α3β1 integrin in preventing tumour cell detachment from the primary tumour in this model (Ref. Reference Bockhorn21). The view that α3β1 can act as a negative regulator of motility gained further support from the recent analysis of α3-null keratinocytes, which displayed enhanced migration in vitro and faster wound healing in vivo (Ref. Reference Margadant22).
In addition to promoting cell–substrate adhesion, the antimigratory activity of α3β1 integrin might involve an ability to promote stable cell–cell junctions. An early study demonstrated that α3 integrin localises to cell–cell contact sites in keratinocytes, and that cell–cell adhesion could be inhibited by an anti-α3 function-blocking antibody (Ref. Reference Carter23). Subsequently, it was shown that expression of α3 integrin, laminin-5, the cell–cell adhesion protein E-cadherin (CDH1) and the gap-junctional protein connexin-43 (GJA1) is coordinately regulated during wound healing, and that α3-dependent keratinocyte adhesion on laminin-5 promotes gap-junction formation (Ref. Reference Lampe24). Concordantly, in kidney epithelial cells from α3-null mice, E-cadherin localisation became less well organised, and other adherens junction components were also displaced, without removal of E-cadherin from the cell surface (Ref. Reference Wang25). The organisation of the cortical actin cytoskeleton also appeared altered, with diminished actin at cell–cell junctions and increased stress fibres at the basal cell surface. Increased stress-fibre formation was also observed in basal keratinocytes in the epidermis of α3-null mice (Ref. Reference Hodivala-Dilke26). Evidence that α3β1 regulates cell junctions in transformed cells comes from studies showing that adhesion on laminin-5 promotes E-cadherin localisation to cell–cell contact sites in HT-29 and Caco-2 colon carcinoma cells (Refs Reference Chartier27, Reference Schreider28). In addition, an anti-α3 function-blocking antibody perturbed the organisation of cell–cell contacts in A431 epidermoid carcinoma cells (Ref. Reference Shigeta29).
The junction-stabilising properties of α3β1-integrin-dependent adhesion on laminin-5 contrast sharply with the junction-destabilising activity reported for the α2β1-integrin ligand collagen I (Ref. Reference Koenig30), or the α5β1 and αvβ3 ligand fibronectin (Ref. Reference Wang31). In a side-by-side comparison, Madin–Darby canine kidney (MDCK) cell scattering triggered by hepatocyte growth factor (HGF) was supported by collagen and fibronectin, but attenuated by laminin (Ref. Reference de Rooij32). The breakdown of adherens junctions triggered by HGF during scattering was found to be a mechanical phenomenon in which the strength of cell–substrate traction forces correlated with the efficiency of adherens junction disruption. The more strongly the cells were able to pull against their substrate, the more able they were to physically pull apart the cell–cell junctions holding them together (Ref. Reference de Rooij32). The reduced scattering observed on laminin may therefore be related to the observations discussed above that cell adhesion on laminin results in fewer actin stress fibres and smaller focal contacts than adhesion on other integrin ligands.
Aside from acting as a negative regulator of cell motility in some settings, α3β1 integrin may also have the potential to suppress cell proliferation at secondary tumour sites. Forced expression of α3β1 was antiproliferative in squamous papillomas resulting from a classic two-stage carcinogenesis protocol (Ref. Reference Owens and Watt33) and in rhabdosarcoma cells implanted subcutaneously (Ref. Reference Weitzman, Hemler and Brodt34). Recent findings that α3β1 can act as a regulator of transforming growth factor β1 (TGF-β1/TGFB1) signalling (Refs Reference Kim35, Reference Reynolds36) could also help to explain the opposing roles of α3β1 in metastatic progression. Like α3β1, TGF-β1 can also display opposing, context-dependent functions in tumour progression and metastasis (Ref. Reference Padua and Massagué37).
In summary, the ability of α3β1 integrin to promote invasion and migration in some tumours may be balanced by its ability to promote ‘stay-at-home’ cell–substrate or cell–cell adhesion or to suppress colonisation at secondary sites in other cases. Whether α3β1's pro- or antimetastatic activities predominate may depend on additional factors such as E-cadherin expression status, passive versus active modes of invasion, or the array of integrin ligands present at primary or secondary tumour sites. As discussed below, the balance between α3β1's opposing roles in metastasis might also be controlled by the complement of α3-associated tetraspanin proteins present in different tumour cell types.
Potential roles of α6β4 and α6β1 integrins in metastasis
In contrast to the α3 integrin subunit, whose expression correlates positively or negatively with metastasis in roughly equal numbers of studies, the expression of the α6 integrin subunit has shown positive correlation in the majority of reports (Table 2). There have also been significantly more in vivo studies addressing the functions of α6 integrins in various models of metastatic cell behaviours (Table 3). Although α6 can pair with either the β1 or β4 subunit, the α6β4 heterodimer has received the most attention by far. Most of the in vivo studies in Table 3 were performed with tumour cell types expressing the α6β4 heterodimer, with the virtually unanimous verdict being that α6β4 integrin has the ability to act as a promoter of tumour progression and metastasis. The unique properties of the β4 integrin cytoplasmic tail may be largely responsible for the α6β4 integrin's prometastatic functions.
Table 2. Clinical or in vivo experimental studies reporting correlations between α6 integrin expression and metastasis

aND indicates that the identity of the β subunit paired with α6 was not determined.
Table 3. In vivo models of metastasis in which α6 integrins were targeted or manipulated

Abbreviations: Ab, antibody; shRNA, short hairpin RNA.
aMDA-MB-435 cells originally believed to be breast carcinoma appear to be melanoma-like.
The structure and function of integrin β4 has been the subject of several excellent recent reviews (Refs Reference Lipscomb and Mercurio38, Reference Giancotti39, Reference Wilhelmsen, Litjens and Sonnenberg40), key points of which are summarised briefly here. The β4 cytoplasmic tail, which unlike those of other β integrin subunits is over 1000 amino acids in length, contains interaction domains for several proteins. These β4-interacting proteins include (1) plectin (PLEC1), which links β4 to the keratin cytoskeleton, (2) the hemidesmosomal proteins BP180 (COL17A1) and BP230 (DST), (3) Src (SRC) family kinases, which phosphorylate multiple tyrosine residues in the β4 tail, (iv) the adaptor protein Shc (SHC), which binds to β4 phosphotyrosines and activates the Ras–MAPK (mitogen-activated protein kinase) pathway, and (5) 14-3-3 proteins, which may help promote lateral association of α6β4 with receptor tyrosine kinases. A primary physiological role of α6β4 is to organise epithelial cell hemidesmosomes by binding laminin-5 in the basement membrane and providing mechanical linkage to the keratin intermediate network. Oncogenic activation of signal transduction pathways can result in tyrosine phosphorylation of the Shc-binding site of β4 as well as protein kinase C (PKC) phosphorylation of key serine residues. Collectively, these modifications result in disassembly of hemidesmosomes and mobilisation of signalling-activated α6β4 integrin. Mobilised α6β4 switches from keratin to actin filament association and may mediate migration and invasion on laminin isoforms. However, many of α6β4's most potent prometastatic functions may involve its ability to act as an amplifier of signalling emanating from activated receptor tyrosine kinases and thereby promote tumour cell survival. These α6β4-mediated prosurvival functions include signalling through phosphoinositide 3-kinase (PI3K) to activate the antiapoptotic serine/threonine kinase Akt (AKT1) and promote the expression of vascular endothelial growth factor (VEGF). VEGF can both stimulate tumour angiogenesis and exert autocrine effects on the tumour cells themselves. Many of these prometastatic functions of α6β4 may occur independently of laminin binding, reflecting the intrinsic signalling capacity of the β4 cytoplasmic tail. However, α6β4's ability to promote tumourigenesis and progression when transgenically expressed in suprabasal keratinocytes, as well as its ability to support the malignant transformation of human keratinocytes, both depended on binding to laminin-5 (Refs Reference Owens41, Reference Dajee42).
The cumulative benefits of mobilised α6β4 integrin for tumour progression may explain why this integrin (which plays a crucial role in the maintenance of epithelial structure and thus might be expected a priori to be a metastasis suppressor) is maintained or even elevated in so many carcinomas. Prostate carcinoma is a notable exception. During prostate cancer progression, the α6β4 ligand laminin-5 and the β4 integrin subunit are frequently both downregulated, with α6 switching to the α6β1 form (Ref. Reference Cress43). The α6β1 integrin binds to laminin-10/11, which is present on the surface of nerves innervating the prostate gland that may serve as a pathway for tumour cell escape from the prostate. Laminin-10/11 is also abundant in the bone matrix, a major site of prostate cancer metastasis, and antibody blockade of α6 integrin can inhibit both bone colonisation and local invasion within the bone (Refs Reference Ports44, Reference King45).
Despite the data implicating it in malignant progression, α6β4 integrin can display tumour-suppressive activities. In p53 (TP53)-deficient, transformed keratinocytes, genetic deletion of α6β4 enhanced tumour formation, growth, and proliferative index (Ref. Reference Raymond46). However, when the cells were further transformed with oncogenic Ras, α6β4 switched roles and became a promoter of tumour growth. The tumour-suppressive activity of α6β4 observed in the absence of oncogenic Ras did not require ligand engagement, but it was abolished by a point mutation that disrupted the interaction of the β4 cytoplasmic tail with the hemidesmosomal protein plectin (Ref. Reference Raymond46). Thus, the loss of β4 expression that occurs during the progression of certain tumour types may be related to the abrogation of a plectin-dependent antiproliferative signal. In addition, as described above for α3β1 integrin, α6β4 can transduce signals that promote the formation of E-cadherin-based adherens junctions, with the potential to restrain tumour cell motility (Refs Reference Schreider28, Reference Hintermann47).
Potential roles of α7β1 integrin in metastasis
Compared with α3 and α6 integrins, relatively little is known about the role of α7β1 integrin in metastasis. Originally identified in melanoma cells (Ref. Reference Kramer48) and myoblasts (Ref. Reference von der Mark49), the α7 integrin has a relatively restricted physiological distribution, with prominent expression in striated and cardiac muscle and certain endothelial and neuronal cell types (Ref. Reference Velling50). Expression of α7 correlates inversely with metastatic potential in melanoma cells, and forced expression of α7 in highly metastatic cells reduced in vitro cell motility, subcutaneous tumour growth, and lung colonisation after tail-vein inoculation (Ref. Reference Ziober51). A recent study has revealed that α7 integrin may act as a tumour suppressor in a variety of tumour cell types (Ref. Reference Ren52). The mechanism underlying the tumour-suppressive activity of α7β1 remains unclear, but may involve upregulation of inhibitors of cyclin-dependent kinases and Rac family small GTPases (Ref. Reference Ren52).
Tetraspanin control mechanisms for laminin-binding integrins
Integrin–tetraspanin complexes
Leaving aside the unique signalling properties of the β4 integrin subunit, a question that remains for the other laminin-binding integrins is how they are able to mediate cellular responses that, as described earlier, may differ from those of other β1 integrins. Part of the answer may lie in the targeting of the laminin-binding integrins to distinct microdomains on the plasma membrane defined by the presence of tetraspanin proteins (tetraspanin-enriched microdomains or TEMs; Fig. 1).
Tetraspanins are a large family of proteins (33 in mammals) that are characterised by four transmembrane domains, cytoplasmic N- and C-termini, and two extracellular domains (EC1 and EC2). A recent high-resolution cryoelectron microscopy structure of the tetraspanin-based urothelial plaques revealed that the smaller EC1 domain packs in under the larger EC2 domain (Ref. Reference Min53), as had been previously predicted by molecular modelling (Ref. Reference Seigneuret54). A unique signature of disulfide bonds and a partially conserved fold within EC2 distinguishes the tetraspanin family from other four-pass proteins (Ref. Reference Seigneuret55). The X-ray crystal structure of the EC2 domain of tetraspanin CD81 suggested that CD81 may exist as a homodimer (Ref. Reference Kitadokoro56). However, the homodimeric interface in the EC2 crystal structure may actually be involved in intramolecular packing of the EC2 and EC1 domains in intact CD81 (Ref. Reference Seigneuret54). Nevertheless, biochemical crosslinking experiments suggest that tetraspanin homodimers can form, and that the homodimeric interface may involve transmembrane domains 1 and 2 (Refs Reference Kovalenko57, Reference Kovalenko58). Tetraspanins heterodimers also occur, as exemplified by the urothelial plaque cryoelectron microscopy data (Ref. Reference Min53). Polar amino acid residues within transmembrane domains 1, 2 and 4, and conserved heptad repeat sequences in transmembrane domains 1–3 are likely involved in inter- and intramolecular packing interactions that support conformational stability, and homotypic and heterotypic tetraspanin–tetraspanin interactions (Refs Reference Kovalenko57, Reference Kovalenko58, Reference Bari59). The simplified schematic in Figure 1 is not meant to imply a fixed stoichiometry; rather, except for specialised cases such as urothelial plaques, TEMs appear to be both molecularly heterogeneous [as indicated by fluorescence deconvolution and immunoelectron microscopy (Ref. Reference Nydegger60)] and highly dynamic [as indicated by single-particle tracking and sophisticated, analytical fluorescence techniques (Refs Reference Barreiro61, Reference Espenel62, and reviewed in Ref. Reference Yanez-Mo63)].
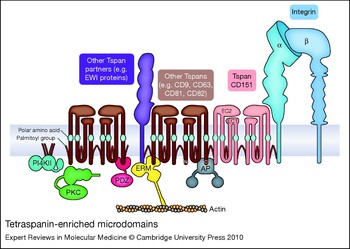
Figure 1. Tetraspanin-enriched microdomains. Laminin-binding integrins associate directly with tetraspanin CD151 and are linked to other tetraspanins (such as CD9, CD63, CD81 and CD82) and tetraspanin partner proteins, such as the immunoglobulin superfamily proteins EWI-2 and EWI-F (the EWI proteins). Cytoplasmic effectors associated with tetraspanin-enriched microdomains (TEMs) include type II phosphatidylinositol 4-kinases (PI4KIIs), classical protein kinase C (PKC) isoforms, ezrin–radixin–moesin family proteins (ERMs), and AP adaptor proteins (APs). Key structural features of tetraspanins include large and small extracellular domains (EC2 and EC1, respectively), polar amino acid residues within transmembrane domains 1, 3 and 4, and palmitoylation of intracellular cysteine residues.
Membrane-proximal, cytoplasmic cysteine residues in tetraspanins are modified by acylation with palmitic acid. These palmitoyl residues play a crucial role in maintaining tetraspanin–tetraspanin associations within TEMs (Refs Reference Berditchevski64, Reference Charrin65, Reference Yang66, Reference Yang67). In addition, integrin subunits α3, α6 and β4 are all palmitoylated, which, at least in the case of the β4 subunit, may contribute somewhat to integrin–tetraspanin interactions (Ref. Reference Yang67). While palmitoylation is crucial for tetraspanin–tetraspanin interactions, the primary way by which laminin-binding integrins are targeted to TEMs is via direct association with tetraspanin CD151. The α3 integrin subunit extracellular domain interacts directly with the EC2 domain of CD151 in an association that can be captured by chemical crosslinking and is stable in strong detergents (Refs Reference Yauch68, Reference Yauch69, Reference Serru70). In some cell types, virtually all the α3 integrin is complexed with CD151 (Ref. Reference Yauch68). The α6 subunit also interacts directly with CD151 (Ref. Reference Kazarov71), but the α6–CD151 complex may be somewhat less stable in many cases (Refs Reference Yauch68, Reference Yauch69). Although not as extensively studied, the association of α7 integrin with CD151 also appears to be a strong, direct protein–protein interaction (Ref. Reference Sterk72). CD151 association with the laminin-binding integrins occurs early in biosynthesis (Refs Reference Kazarov71, Reference Berditchevski73); thus, CD151 and its integrin partners most likely traffic together to the cell surface. Once there, CD151 links the laminin-binding integrins to other tetraspanins, such as CD9 and CD81. These other tetraspanins may in turn have their own major partners (Fig. 1). For example, CD9 and CD81 associate with two related immunoglobin superfamily (IgSF) proteins – EWI-2 (PGRL/CD316/IGSF8) and EWI-F (CD9P-1/FPRP/PTGFRN) – and link them via CD151 to α3β1 integrin (Refs Reference Stipp, Kolesnikova and Hemler74, Reference Charrin75). As predicted by the simplified working model in Figure 1, genetic ablation or silencing of CD151 by RNA interference (RNAi) largely disrupts the association of the laminin-binding integrins with other TEM-resident proteins such as CD9 and CD81 (Refs Reference Winterwood12, Reference Yang76, Reference Takeda77, Reference Yamada78), although some residual CD151-independent associations may remain.
The EWI proteins serve only as representative tetraspanin partners in Figure 1. Proteomic and biochemical analyses have revealed numerous other TEM-resident proteins, including (1) other IgSF proteins, such as CD19 and MHC class I antigen, (2) cell-surface proteases such as ADAM10, (3) membrane-bound growth factors, such as HB-EGF (HBEGF), (4) growth factor receptors, such as the EGF receptor (EGFR), and (5) other multitransmembrane-domain proteins such as claudin 1 (CLDN1), the heterotrimeric G-protein-coupled receptor GPR56, and putative choline transporters CTL1 (SLC44A1) and CTL2 (SLC44A2) (Refs Reference André79, Reference Le Naour80, Reference Kovalenko, Yang and Hemler81; see also the reviews Refs Reference Hemler82, Reference Levy and Shoham83). Thus, the targeting of laminin-binding integrins to TEMs by CD151 creates many opportunities for functional collaborations between the integrins and other cell-surface proteins, or for the functional modification of integrin-mediated responses.
While the majority of TEM-resident proteins identified thus far are transmembrane proteins, some cytoplasmic components have been identified as well (Fig. 1). Classical PKC isoforms PCKα and PCKβ (PRKCA and B) associate with tetraspanins upon activation and are linked via tetraspanins to laminin-binding integrins (Ref. Reference Zhang, Bontrager and Hemler84). Type II phosphatidylinositol 4-kinases (PI4KIIA and B) also associate with specific tetraspanins and are thereby linked to laminin-binding integrins (Refs Reference Yauch68, Reference Berditchevski85, Reference Yauch and Hemler86). Actin-binding ezrin–radixin–moesin (ERM) family proteins may interact with the cytoplasmic tails of CD81 and the CD81 partner proteins EWI-2 and EWI-F, potentially creating a link between TEMs and the actin cytoskeleton (Refs Reference Chang and Finnemann87, Reference Sala-Valdes88). Several tetraspanins possess potential tyrosine-based sorting motifs in their C-termini (Refs Reference Stipp, Kolesnikova and Hemler89, Reference Berditchevski and Odintsova90). These motifs (of the form YXXΦ, where Y represents tyrosine, and Φ represents a hydrophobic amino acid) are recognised by AP adaptor complexes that link cargo to clathrin in clathrin-coated vesicles. The YXXΦ motif in tetraspanin CD63 is recognised by the μ subunit found in the AP-3 (and to a lesser extent AP-2) complex (Ref. Reference Rous91), and mutations in the motif alter the subcellular localisation of CD63. Syntenin-1 (SDCBP), a PDZ-domain-containing protein, also binds to the C-terminus of CD63 and inhibits the AP-2-dependent internalisation of CD63 from the cell surface (Ref. Reference Latysheva92). In addition, the PDZ-domain proteins EBP50 (SLC9A3R1) and Sap97 (DLG1) bind to the CD81 C-terminal tail (Ref. Reference Pan93). Several other tetraspanins possess potential PDZ-domain-binding sites, so it will be important to determine if other PDZ-domain proteins also localise to TEMs. Lastly, consistent with the association of heterotrimeric G-protein-coupled receptors with tetraspanins, heterotrimeric G proteins themselves have also been detected in tetraspanin complexes (Refs Reference Le Naour80, Reference Little, Hemler and Stipp94) (not shown in Fig. 1).
Numerous studies indicate that the localisation of laminin-binding integrins to TEMs is functionally relevant. Antitetraspanin antibodies, forced expression of tetraspanin mutants, genetic deletion, and RNAi-mediated silencing of tetraspanin expression have all produced examples in which the function of laminin-binding integrins has been compromised (see Refs Reference Hemler82, Reference Charrin95, Reference Berditchevski96). Nevertheless, the molecular mechanisms by which tetraspanins regulate integrin function remain murky. The sections below highlight potential tetraspanin control mechanisms for laminin-binding integrins that have emerged from recent studies.
Do tetraspanins regulate integrin–ligand interactions?
Two studies have proposed that tetraspanins may regulate integrin–ligand binding by regulating integrin affinity state. In one study, a soluble α5β1-integrin-ligand mimetic (RGD peptide) was added to parental Chinese hamster ovary (CHO) cells or to a CHO cell subclone overexpressing CD9 (Ref. Reference Kotha97). The binding of an anti-β1-integrin antibody that recognises a ligand-induced binding site (LIBS) epitope was then assessed by flow cytometry. The apparent upregulation of the LIBS epitope on the CD9-overexpressing cells in response to RGD was modestly higher than that of the parental CHO cells. However, because the CD9–α5β1 association can be observed only in very mild detergents (Ref. Reference Fitter98), it may be indirect, raising the question of how CD9 could directly modulate the α5β1 affinity state. An alternative explanation is that CD9 expression could change the clustering or organisation of α5β1 on CHO cells thereby improving the avidity of the bivalent LIBS antibody used in this study.
In the other study, a preparation of α3β1 integrin apparently depleted of associated CD151 was compared with non-CD151-depleted α3β1 upon reconstitution into liposomes (Ref. Reference Nishiuchi99). The CD151-depleted α3β1 liposomes bound somewhat less well to laminin-10/11 compared with nondepleted liposomes. The CD151-depleted liposomes also showed reduced binding to wells coated with an anti-β1-integrin-LIBS antibody. Adding back CD151 to the CD151-depleted liposomes restored their binding to laminin-coated wells to normal levels. These results were interpreted as evidence that CD151 might stabilise the activated, ligand-bound conformation of α3β1 integrin, but other interpretations are possible. Given the ability of tetraspanin proteins to self-associate, increasing CD151 content in liposomes might create clusters or patches containing higher densities of α3β1, enhancing its avidity for physiological or nonphysiological ligands. This issue might not be fully resolved until monovalent, soluble ligand mimetics are available that can detect preconfigured, activated laminin-binding integrins in the way that, for example, Fab fragments of the WOW-1 antibody can detect activated β3 integrins (Ref. Reference Pampori100). Of potential relevance, CD151 regulates post-ligand-binding signalling events mediated by αIIbβ3 integrin in platelets without influencing ligand binding itself (Ref. Reference Lau101). Thus, at least in this case, it appears that CD151 does not act by regulating the affinity state of its integrin partner.
Some studies have reported that genetic deletion or RNAi-mediated silencing of CD151 results in reduced adhesion on laminin isoforms (Refs Reference Winterwood12, Reference Yamada78, Reference Nishiuchi99, Reference Geary102, Reference Hasegawa103). However, others have reported no difference in short-term adhesion assays for CD151-depleted cells (Ref. Reference Takeda77). Reduced adhesion should not be construed as evidence for reduced integrin–ligand binding affinity. Adhesion differences in such assays more likely reflect impaired kinetics of cell spreading on laminin, which is a generally agreed outcome of silencing or deleting CD151 (Refs Reference Winterwood12, Reference Takeda77, Reference Geary102, Reference Hasegawa103). Whether or not a difference in adhesion is observed may depend upon detachment force or ligand density, which may vary from one study to another.
Spreading defects in CD151-silenced cells may be related to the role of CD151 in promoting adhesion strengthening – the increase in resistance to detachment that follows initial adhesion (Ref. Reference Lammerding104). The mechanisms of adhesion strengthening are unclear but may involve several factors, including a progressively increasing area of cell–substrate attachment, recruitment of additional integrins to attachment sites, force-dependent integrin activation, and increased strength of the integrin–cytoskeleton interaction such that membrane stiffness in the vicinity of ligated integrins increases (Refs Reference Gallant, Michael and Garcia105, Reference Puklin-Faucher and Sheetz106). The potential for TEMs to provide auxiliary links to the actin cytoskeleton via ERM proteins or scaffolds organised by PDZ-domain proteins may thus be of relevance (Fig. 1). In this regard, it is interesting to note that a CD151 C-terminal cytoplasmic tail mutant was defective in its ability to promote α6β1-integrin-dependent adhesion strengthening (Ref. Reference Lammerding104). Tetraspanin association might also facilitate the recruitment of laminin-binding integrins into adhesive nanoclusters, as has been described for the cell adhesion molecules VCAM and ICAM, which associate with tetraspanins in endothelial cells (Ref. Reference Barreiro61). A similar role has been proposed for tetraspanin CD81, which promotes adhesion strengthening mediated by its partner α4β1 integrin under shear-flow conditions (Ref. Reference Feigelson107).
Tetraspanin regulation of integrin trafficking
A variety of studies show that tetraspanins can regulate the trafficking of their partners (reviewed in Refs Reference Berditchevski and Odintsova90, Reference Charrin95). In CD151-silenced carcinoma cells migrating on laminin-5, the rate of α3 integrin internalisation was significantly impaired compared with wild-type cells (Ref. Reference Winterwood12). Concurrently, persistent protrusions formed at lateral and trailing edges of the migrating tumour cells. Since continuous forward migration depends not only on the formation of new adhesive contacts at the front of the cell but also the disassembly of old adhesive contacts at the rear, one way that tetraspanin association might facilitate the rapid migration mediated by laminin-binding integrins is by promoting efficient integrin internalisation at the lateral or trailing edges of migrating cells. Different complements of tetraspanins might influence integrin trafficking in different cell types. For example, enforced expression of tetraspanin CD82 in Du145 prostate carcinoma cells reduced α6 integrin cell-surface expression, enhanced ligand-induced α6 internalisation, and impaired α6-dependent adhesion and morphogenesis (Ref. Reference He108). However, cell-surface α6 was not altered upon CD82 expression in PC-3 prostate carcinoma cells (Ref. Reference Sridhar and Miranti109), so the effect of CD82 on integrin cell-surface expression is different in different cell types. Mutation of the YXXΦ motif in the CD151 C-terminal cytoplasmic tail strongly inhibited CD151 internalisation from the cell surface, suggesting that this motif, which might interact with AP adaptor proteins (Fig. 1), could be important for regulating integrin trafficking (Ref. Reference Liu110). However, no difference in the internalisation rate of total cell-surface α6 integrin was observed in cells overexpressing the CD151 YXXΦ mutant, so the magnitude of its impact on integrin trafficking remains to be determined. In addition, the CD82 YXXΦ motif appears not to mediate an association with the AP-2 adaptor complex, and may not be required for CD82 internalisation (Ref. Reference Xu111). Thus, the functionality of the putative YXXΦ motifs is different for different tetraspanins.
Tetraspanin regulation of integrin signalling
In several independent studies, RNAi-mediated silencing or genetic deletion of tetraspanin CD151 has resulted in impaired signalling through laminin-binding integrins (Refs Reference Yang76, Reference Takeda77, Reference Yamada78, Reference Sadej112). Signalling molecules whose activities were reduced in CD151-deficient cells include tyrosine kinases FAK, Src and Lck (LCK), serine/threonine kinases Akt and ERK (MAPK1), and small GTPases Rac1 and Cdc42. Reduced activation of nitric oxide synthase (eNOS, NOS3) and reduced phosphorylation of the focal adhesion scaffold protein paxillin (PXN) have also been reported. In addition, enforced CD82 expression inhibited integrin-dependent crosstalk with the c-Met (MET) receptor tyrosine kinase and Src signalling through the Src substrates FAK and p130CAS, an adaptor protein important for integrin-driven cell motility (Ref. Reference Sridhar and Miranti109). CD82 expression might also influence the outright expression level of p130CAS and thus p130CAS-dependent signalling (Ref. Reference Zhang113).
Collectively, these data indicate that tetraspanin association makes critical contributions to signalling through laminin-binding integrins. However, it remains to be established whether tetraspanin association is simply required for laminin-binding integrins to signal properly or whether tetraspanins endow laminin-binding integrins with some of the unique properties that distinguish them from other integrins. As discussed in preceding sections, cells adhering on laminins may display less activation of the small GTPase RhoA, fewer actin stress fibres, smaller focal adhesions, and enhanced stability of cell–cell junctions, compared with cells adhering on other integrin ligands. Do tetraspanins contribute to any of these special properties? Support for the possibility that they do comes from a recent study of A431 epidermoid carcinoma cells, which deposit and adhere to their own laminin-5-rich matrix and normally display relatively orderly cell–cell junctions. Near-total RNAi-mediated silencing of CD151 in A431 cells resulted in significantly elevated RhoA activity, increased actin stress fibres, disorganised adherens junctions, and a more highly dynamic monolayer with a reduced lifespan of cell–cell contacts (Ref. Reference Johnson114). Treating cells with a cell-permeable RhoA inhibitor or re-expressing wild-type CD151 in the silenced cells reversed these phenotypes, but re-expressing a CD151 mutant with impaired α3β1 integrin association did not. These data suggest that upon loss of CD151 association, α3β1 may lose some of the unique functions that set it apart from the non-laminin-binding integrins. Supporting these observations, silencing CD151 in HSC5 epidermal carcinoma cells or in embryonic kidney cells also resulted in elevated stress-fibre formation (Refs Reference Hasegawa103, Reference Chattopadhyay115), and forced expression of a CD151 palmitoylation mutant disrupted α3β1 integrin association with other tetraspanins and increased the size of focal adhesions in Rat-1 fibroblasts plated on laminin-5 (Ref. Reference Sawada116). In addition, an α3 mutant with impaired association with CD151 failed to rescue the disturbed cell–cell junctions observed in α3-null kidney epithelial cells (Ref. Reference Chattopadhyay115).
Figure 2 depicts a working model based on the observations above. Laminin-binding integrins associate with tetraspanin CD151 early in biosynthesis, resulting in delivery of the integrins to TEMs on the cell surface. Tetraspanin association may allow laminin-binding integrins to organise smaller, more-promigratory focal contacts than other integrins. Tetraspanins also contribute to signalling events required for rapid cell migration and may facilitate disassembly and recycling of old adhesive contacts. Collectively, these functions support rapid tumour cell migration, invasion and metastasis. Balancing these promigratory functions, tetraspanin association may also allow the laminin-binding integrins either to generate a Rho-suppressive signal or to avoid activating Rho to the same extent as other integrins, resulting in a low basal level of Rho activity compatible with stable adherens junctions. E-cadherin itself may also participate in these Rho-suppressive functions (Ref. Reference Noren117). Stable cell–cell junctions may function to oppose tumour cell migration and invasion. Such an arrangement could help to explain the divided literature on the roles of laminin-binding integrins in metastasis. Depending on which aspect of their tetraspanin-controlled functions predominates in a particular setting, laminin-binding integrins could function as either promoters or suppressors of metastasis. The tetraspanin expression profiles in different cell types could help determine which functions predominate. For example, tetraspanins CD9 and CD82 have been implicated both as promoters of junctional organisation (Refs Reference Furuya118, Reference Abe119) and as suppressors of tumour cell motility (Refs Reference Zoller120, Reference Miranti121). Thus, CD9 and CD82 may tilt the scale of laminin-binding integrin function towards the metastasis-suppressive side, possibly helping to explain why these tetraspanins have frequently been implicated as metastasis suppressors (Refs Reference Zoller120, Reference Miranti121). The unique properties of α6β4 integrin, both in terms of its ability to organise hemidesmosomes in stable epithelia and in terms of the potent, β4-dependent, prometastatic signalling functions that can be unleashed during carcinoma progression, may not fit neatly into the working model shown in Figure 2. However, CD151 association is known to contribute to α6β4's promigratory functions (Ref. Reference Yang76), and has been proposed to contribute to its hemidesmosomal functions as well (Ref. Reference Sterk122).

Figure 2. Consequences of TEM localisation for laminin-binding integrins. Laminin-binding integrins (represented by α3β1) associate with tetraspanin CD151 via a direct protein–protein interaction between the α integrin subunit ectodomain and the CD151 EC2 domain. The integrin–CD151 complex forms early in biosynthesis and is delivered to cell-surface tetraspanin-enriched microdomains (TEMs). TEM localisation may promote α3β1-dependent tumour cell migration on laminin-5 in multiple ways including (1) facilitating the formation of small, promigratory focal contacts, (2) participating in promigratory signalling, and (3) facilitating efficient internalisation of α3β1 at the trailing edge of migrating cells. TEM localisation may also allow α3β1 to collaborate with E-cadherin to reduce the activity of the RhoA small GTPase to a low basal level. Too much RhoA activity can destabilise adherens junctions, which normally function to restrain tumour cell motility. Thus, by keeping RhoA activity low, α3β1–tetraspanin complexes might exert an antimigratory activity that balances α3β1's promigratory functions.
Possible clinical implications and outstanding questions
Laminin-binding integrins and their tetraspanin partners might be targeted by a variety of approaches including function-modulating monoclonal antibodies, RNAi-based therapeutics, and, in the case of tetraspanin proteins, recombinant soluble EC2-domain mimics. Current prospects for development of these different interventions to the stage of clinical trials have been recently reviewed elsewhere (Ref. Reference Hemler123). The discussion here focuses on gaps in our knowledge that need to be filled to support the clinical use of reagents targeting laminin-binding integrins and tetraspanins, as more such reagents become available.
Should the laminin-binding integrins be directly targeted?
The laminin-binding integrins have important developmental and tissue-maintenance functions, so the possibility of directly targeting them in cancer with function-blocking or expression-blocking therapeutics will likely depend upon the ability to deliver the therapeutic specifically to tumour cells. Assuming that this technical hurdle can eventually be overcome, the question remains ‘under what circumstances would it be advantageous to extinguish laminin-binding integrin function in tumour cells?’ Thus far, available data on α6β4 integrin from in vivo models (Table 3) suggest that for tumours bearing signalling-activated, mobilised α6β4 integrin, blocking its function would in most cases be beneficial. There may be exceptions, such as certain gastric carcinomas, in which α6β4 may play a suppressive role (Table 2). In addition, analysis of α6β4 in transformed keratinocytes suggests that blocking its function in early-stage tumours might in some cases be counterproductive (Ref. Reference Raymond46). For α3β1 integrin, the situation may be more complex. As the studies summarised in Table 1 and the working model in Figure 2 both emphasise, α3β1's function in tumour progression is likely to be highly dependent on context. For example, in situations where α3β1 is making a strong contribution to the maintenance of stable cell–cell junctions, blocking its function might result in a more invasive phenotype rather than reduced invasion. Thus, the role of α3β1 may depend upon factors such as E-cadherin expression status or the complement of tetraspanin proteins present in specific tumours. For α7β1 integrin, the relatively limited data available indicate that it plays a tumour-suppressive role in multiple tumour cell types (Ref. Reference Ren52).
An alternative to blocking or silencing laminin-binding integrins would be to modify their function. Thus far, no α-integrin-subunit-specific activating antibodies have been reported; however, an antimetastatic α6 integrin antibody that inhibits regulated ectodomain cleavage of the α6 subunit (but not α6-dependent adhesion) has recently been described (Ref. Reference Ports44). In addition, an anti-α3-integrin antibody that does not block adhesion has been reported to block keratinocyte migration on laminin-5 (Ref. Reference Hintermann124). It might be beneficial to re-evaluate the activities of the wealth of available anti-integrin antibodies, in light of the more sophisticated knowledge of integrin function that has accumulated since many of these reagents were first generated. The numerous associations of the β4 integrin subunit with receptor tyrosine kinases and intracellular signalling proteins might create the potential for targeting β4 signalling functions without disrupting β4-dependent adhesion (Ref. Reference Giancotti39). Mice bearing a β4 subunit with impaired signalling functions are viable and fertile (Ref. Reference Nikolopoulos125), suggesting that inhibitors of β4 signalling might not disrupt critical physiological functions.
Prospects for modulating laminin-binding integrin function by targeting TEMs
Since its identification as a major, directly interacting partner of the laminin-binding integrins, CD151 has received significant attention as a potential therapeutic target in cancer. Increased CD151 expression correlates with increased risk of metastasis or reduced survival in colon, lung and prostate cancers (Refs Reference Tokuhara126, Reference Hashida127, Reference Ang128). Elevated CD151 expression is also observed in some breast cancers and correlates with reduced survival in breast cancer patients (Refs Reference Yang76, Reference Sadej112). RNAi-mediated silencing of CD151 slowed the growth of orthotopic or subcutaneously xenografted MDA-MB-231 breast carcinoma cells (Refs Reference Yang76, Reference Sadej112). In addition, an anti-CD151 monoclonal antibody strongly inhibited spontaneous metastasis in a chick embryo xenograft model (Ref. Reference Zijlstra129). Collectively, these studies have established CD151 as a potentially attractive target; however, several important questions remain. First, while it is clear that loss of CD151 can impair the functions of laminin-binding integrins, it is not yet known whether this is the basis of the reduced in vivo growth rate of CD151-silenced tumour cells. Similarly, the antimetastatic CD151 antibody inhibits tumour cell intravasation by suppressing cell–matrix detachment at the trailing edge of migrating cells (Ref. Reference Zijlstra129), but it is not clear which integrins are involved in this effect. In addition, kidney failure in human patients and mice lacking CD151 suggests that, as with the laminin-binding integrins themselves, targeting CD151 may require tumour-specific delivery of the therapeutic agent (Refs Reference Sachs130, Reference Karamatic Crew131, Reference Baleato132). Lastly, if the working model in Figure 2 holds, additional factors, such as E-cadherin expression status, may determine in which cases CD151 targeting may be beneficial. In this regard, the MDA-MB-231 tumour cell line, which is the only tumour cell type used thus far for in vivo tumour growth assays after CD151 silencing, lacks E-cadherin expression. Additional experiments with E-cadherin-positive tumour cells may provide important information about the diversity of CD151's functions in tumour progression. The recent identification of DHHC2 (ZDHHC2), an enzyme responsible for palmitoylation of the CD151 (and CD9) cytoplasmic tail, may represent another way in which CD151 function could be modulated (Ref. Reference Sharma, Yang and Hemler133). However, RNAi inhibition of DHHC2 disrupted E-cadherin-based carcinoma cell–cell junctions (Ref. Reference Sharma, Yang and Hemler133). If the loss of junctions in DHHC2-depeleted cells turns out to be a tetraspanin-dependent phenomenon, it would serve to further underscore the complex roles of integrin–tetraspanin complexes in regulating tumour cell–cell and cell–substrate interactions.
Like CD151, TSPAN8 (CO-029) has been implicated as a promoter of metastasis, and part of TSPAN8's prometastatic functions may come from its ability to associate with α6β4 integrin via CD151 and promote α6β4-dependent migration and metastasis (Ref. Reference Zoller120). TSPAN8 plays an important role in pancreas development in Xenopus embryos (Ref. Reference Jarikji134), but its functions in mammalian development and adult physiology are not well defined, so the extent to which targeting TSPAN8 in tumours might cause undesirable side effects in normal tissues remains unknown.
An alternative to inhibiting the tumour-promoting functions of tetraspanins such as CD151 and TSPAN8 would be to somehow take advantage of the tumour-suppressive functions of tetraspanins such as CD9 and CD82. Proof of principle for such an approach comes from the demonstration that intratracheally instilled adenoviral vectors encoding CD9 or CD82 could suppress lymph-node metastases of orthotopically implanted Lewis lung carcinoma cells (Ref. Reference Takeda135). Since many tumours might not be as amenable to adenoviral delivery, another approach would be to identify small-molecule therapeutics that promote CD9 or CD82 expression. For example, CD82 is targeted for degradation by the ubiquitin E3 ligase gp78 (AMFR), and suppressing gp78 expression by RNAi enhanced CD82 expression and blocked pulmonary metastasis of fibrosarcoma cells (Ref. Reference Tsai136). Thus, selective small-molecule inhibitors that target specific components of the ubiquitin–proteasome system, such as individual E3 ligases (Ref. Reference Guédat and Colland137), might be screened for their effects on tetraspanin expression. Lastly, while there is significant evidence that CD9 and CD82 can both influence cell motility, it still remains unclear whether this is the primary mode by which they exert antimetastatic effects (Refs Reference Zoller120, Reference Miranti121). More structure–function studies coupled with in vivo metastatic colonisation assays are needed to address this question.
Conclusion
The laminin-binding integrins are potential therapeutic targets for inhibition of metastasis, but they also fulfil critical physiological functions in the organisation and maintenance of epithelial cell layers. Tetraspanin proteins may offer the potential to modulate the functions of laminin-binding integrins in more subtle ways, with less likelihood of disrupting key physiological functions. Important outstanding questions include defining in which settings the laminin-binding integrins are acting as promoters versus suppressors of metastasis, and clarifying the molecular mechanisms by which tetraspanins regulate integrin function. Advances in the clinical use of RNAi and the development of small-molecule inhibitors that can modulate tetraspanin expression or function may bring with them the promise of finally unlocking the therapeutic potential of the laminin-binding integrins.
Acknowledgements and funding
The author gratefully acknowledges insightful comments from the reviewers of this manuscript, as well as grant support from the Roy J. Carver Charitable Trust (07-2869), the American Cancer Society (RSG-07-043-01-CSM), and the National Institutes of Health (CA136664). Mary E. Herndon provided a helpful critique of the manuscript, and members of the University of Iowa Tumor Biology & Genetics Group provided helpful feedback on many of the ideas presented here.






 Open access
Open access