


Retrospective associations between low weight or thinness at birth and the risk of CVD(Reference Barker1) and type 2 diabetes(Reference Barker, Hales and Fall2) gave rise to the hypothesis that maternal nutritional status may be one of a number of factors that programme the long-term risk of disease(Reference Eriksson, Forsén and Tuomilehto3). This hypothesis has received strong support from studies of small and large animal species, which overwhelmingly indicate that exposure to undernutrition during pregnancy, whether specific to macronutrients (for example, protein)(Reference Bellinger, Sculley and Langley-Evans4–Reference Langley-Evans and Nwagwu6) or micronutrients (for example, Fe)(Reference Gambling, Dunford and Wallace7), or in the form of lower overall food intake(Reference Vickers, Breier and Cutfield8), programmes the risk of adult hypertension, glucose intolerance, insulin resistance and dyslipidaemia(Reference Langley-Evans9).
While there is an extensive literature on the long-term programming effects of maternal undernutrition, the capacity for overnutrition to exert similar programming influences is relatively unexplored. Understanding the impact of maternal obesity is of major importance as the prevalence of obesity is rising rapidly among young women(10). In addition to being a major risk factor for adverse outcomes of pregnancy(10–Reference Walsh13), higher maternal BMI during pregnancy is associated with the risk of obesity in the offspring in later life. A number of recent studies(Reference Calvert, Lefer and Gundewar14–Reference Shankar, Harrell and Liu17) of rodent species have also shown that maternal high-fat feeding during pregnancy can have similar postnatal consequences to undernutrition. Samuelsson et al. (Reference Samuelsson, Matthews and Argenton18), for example, reported that the offspring of mice rendered obese by feeding a high-fat–high-sugar diet for 6 weeks before mating and then throughout pregnancy and lactation were obese, hypertensive and glucose intolerant. Bayol et al. (Reference Bayol, Simbi and Bertrand19) showed that a cafeteria diet protocol introduced at the point of conception in rats could programme adiposity and feeding behaviour in the rat.
In most of the previous literature considering the potential for maternal obesity to programme later disease risk, it has been impossible to assess whether the observed phenotypes in the offspring are a consequence of maternal obesity per se or of other aspects of the obesity-inducing diet (for example, lower protein or higher fat content). The distinction is important as we have previously demonstrated, using the feeding of a maternal cafeteria diet either before or during pregnancy, that fetal growth retardation in rats occurred as a result of maternal obesity and not by the feeding of an energy-dense diet(Reference Akyol, Langley-Evans and McMullen20). Cafeteria feeding provides a useful alternative to the feeding of purified high-fat diets to induce obesity. It avoids the use of very high intakes of a particular type or source of fat while inducing persistent hyperphagia and increased energy intake(Reference Shafat, Murray and Rumsey21, Reference Rothwell and Stock22) as a result of the variety and novelty of the foods available. Although it produces some variation in foods and nutrients consumed between animals in the same group, this approach has been selected as it is more closely aligned with dietary patterns observed in human subjects than conventional purified high-fat diets. The use of dietary patterns as a measure of exposure in human studies of diet–disease relationships is known to provide a stronger basis for any inferred effects than considering specific nutrients.
The aim of the present study was to explore whether the effects of maternal obesity upon fetal growth were associated with a greater risk of metabolic disease in later life. Using a comprehensive experimental design, we addressed the specific hypotheses that exposure to maternal cafeteria feeding in early development would result in insulin resistance during adult life. The study further aimed to assess whether programming effects of maternal cafeteria feeding were dependent upon offspring diet and sex.
Methods
Animals and diets
The experiments were performed under licence from the Home Office in accordance with the 1986 Animals (Scientific Procedures) Act. All animals were housed individually in plastic cages and subjected to a 12 h light–12 h dark cycle at a temperature of 20–22°C and 45 % humidity. The animals were housed on woodshavings and had ad libitum access to food and water at all times. Virgin female Wistar rats (aged 3 weeks; n 64) were randomly allocated to be fed either a control chow diet alone (C, n 29) or a control chow diet alongside a random selection of highly energetic and palatable human foods (cafeteria diet, O; n 35), as described previously by Akyol et al. (Reference Akyol, Langley-Evans and McMullen20). The range of foods offered to cafeteria diet-fed rats included biscuits, potato crisps, fruit and nut chocolate, Mars bars, cheddar cheese, golden syrup cake, pork pie, cocktail sausages, liver and bacon pâté, strawberry jam and peanuts. Each day, four of the cafeteria foods were provided in a bowl on the cage floor, in excess quantities. The foods provided were altered daily, to maintain variety, by replacing two of the foods with new items. Hence, the animals did not receive the same foods for more than two consecutive days at a time. The chow diet (Teklad Global 18 % Protein Rodent Diet; Harlan, Belton) and cafeteria diet foods were individually weighed in and out of the cage between 09.00 and 10.00 hours daily to monitor intake, enabling the calculation of energy and macronutrient intake, after allowing for weight changes due to drying of foods, as described previously(Reference Akyol, Langley-Evans and McMullen20).
The diets were introduced to the females from weaning to allow a sufficient period of cafeteria feeding to induce obesity before mating at 10 weeks of age. After 7 weeks of control or cafeteria feeding, all rats were paired with Wistar stud males, and mating was confirmed by the appearance of a semen plug. In order to separate the effects of maternal cafeteria feeding from the effects of maternal obesity, some of the animals from the control group were randomly allocated to the cafeteria diet (n 18) and some of the animals from the cafeteria group were randomly allocated to the control diet (n 20) on confirmation of mating. The remaining animals within each group were maintained on their pre-gestational diets (C, n 11 and O, n 15). At birth, all litters were culled to a maximum of eight pups (four males and four females, where possible, randomly selected). During lactation, each group was again sub-divided to give chow- and cafeteria diet-fed litters. The overall design for the study is shown in Fig. 1. Each three-letter group name indicates at which stage the rats received either the control diet (C) or cafeteria diet (O). So, for example, group CCO received the control diet during both the pre-mating and pregnancy periods and the cafeteria diet during the lactation period. All litters were weaned at 3 weeks of age. Half of the offspring from each litter were weaned onto the standard chow diet and the remaining weanlings were allocated to receive the chow and cafeteria diets. The protocol for feeding these animals was identical for that used for the mothers (described earlier).

Fig. 1 Study design. Control diet (C, □); cafeteria diet (O, ■). Values for n show the number of successful pregnancies in each group.
Glucose tolerance test
At the age of 13 weeks, all offspring were subject to an intraperitoneal glucose tolerance test(Reference González-Yanes, Serrano and Bermúdez-Silva23). At 18 h before testing, all food was removed from the animals. At the start of the test, the animals were restrained to obtain a baseline blood sample from the superficial tail vein, under local anaesthesia. Within 5 min of sampling, 1 ml/100 g body weight glucose (20 g/100 ml in 0·9 % saline) was administered via an intraperitoneal injection (overall dose of 2 g glucose/kg body weight). Blood was repeatedly sampled from the tail vein at 5, 15, 30 and 60 min post-glucose administration. All blood samples were collected into heparinised capillary tubes and stored on ice until centrifuged in a haematocrit centrifuge. Plasma was collected and stored at − 80°C until required for analysis. Plasma glucose was assayed using an adapted protocol based on the glucose oxidase method(Reference Trinder24). Data on the area under the curve (AUC) for glucose were obtained using GraphPad Prism version 5 (Graphpad Software Inc., La Jolla, CA, USA). The homeostasis model assessment-insulin resistance (HOMA-IR) index was calculated from fasting plasma glucose and insulin concentrations according to the following equation: insulin (μU/ml) × glucose (mg/l)/405. At the end of the glucose tolerance test, the animals were culled using CO2 asphyxia and cervical dislocation. A final blood sample (120 min after administration of glucose) was taken by cardiac puncture and major organs were weighed and snap-frozen in liquid N2.
Metabolic indices
Total plasma cholesterol and total TAG were assayed using commercially available kits (Thermo Life Sciences, Basingstoke, UK). Plasma insulin concentrations were determined at baseline and 30 min using an ELISA kit (Crystal Chem, Inc., Downers Grove, IL, USA), following the manufacturer's instructions.
Determination of mRNA expression
RNA was extracted from snap-frozen liver tissue by the TRIzol procedure (Invitrogen, Paisley, UK) and subjected to DNase treatment (Promega, Southampton, UK), phenol–chloroform extraction and ethanol precipitation(Reference McMullen and Langley-Evans25). RNA was reverse transcribed using Moloney murine leukaemia virus RT (Promega). Real-time PCR primers were designed for insulin receptor substrate 2 (Irs2) and the serine threonine protein kinase, Akt2, using Primer Express software (version 1.5; Applied Biosystems, Paisley, UK) from the DNA sequence GenBank accession numbers NM_001168633 (Irs2) and DQ198085 (Akt2), respectively. The primer sequences were as follows: Irs2, forward 5′-CAAGAACCTGACCGGTGTATACC-3′ and reverse 5′-GGCTGTTCGCAATTGAGCTT-3′; Akt2, forward 5′- CAGAGAGCCGAGTCCTACAGAATAC-3′ and reverse 5′-GTCATGGGTCTGGAAGGCATA-3′. The primers were ordered from MWG Biotech (London, UK). The primer sequences for the housekeeping gene (β-actin) have been published elsewhere(Reference McMullen and Langley-Evans25). Real-time PCR was performed using a LightCycler 480 PCR machine (Roche, Burgess Hill, UK) and SYBR Green Probe (Roche). Expression of Irs2 and Akt2 was normalised to the housekeeping gene, expression of which was unaltered in response to the dietary treatments.
Statistical analysis
All data were analysed using the Statistical Package for Social Sciences (version 16; SPSS, Inc., Chicago, IL, USA). The effect of the pre-gestational and gestational diets on maternal outcomes was assessed using a general linear model three-way ANOVA (fixed factors, maternal diet at two stages and sex). Where longitudinal data were available (for example, weekly body weights), the week of study was used in a repeated-measures analysis. For offspring data, the dataset was split to consider the effects of the cafeteria diet post-weaning separately and the data were analysed using four-way ANOVA (fixed factors, maternal diet at three stages and sex). No more than one male and one female from each litter were included in the analyses. Post hoc testing (Tukey's test) was applied for the main effects of the diet at each stage, but not for the interactions between the exposure periods. Values are expressed as means with their standard errors. P < 0·05 was considered statistically significant. The study was adequately powered to meet the stated aim.
Results
As shown in Table 1, the body weights of the mothers did not vary significantly at the start of the experiment. Over the period before mating, weight gain was greater in all groups of rats that were fed the cafeteria diet during the pre-pregnancy period (effect of pre-pregnancy diet, P < 0·001). The introduction of a cafeteria diet from mating led to greater body weight by the end of pregnancy, and the mothers pre-fed the cafeteria diet before mating remained heavier than the control mothers (P = 0·02). The main influence on weight gain during pregnancy was the maternal diet during pregnancy (P < 0·001). Litter size did not vary significantly between the groups (data not shown). Fig. 2 shows the energy and macronutrient intake of rats during the pre-gestation, pregnancy and lactation periods. As shown in Fig. 2(A) and (B), energy intake was increased during all periods of cafeteria feeding. At points of transition where cafeteria diet-fed rats transferred to the chow diet or chow diet-fed rats transferred to the cafeteria diet, we found no evidence of acute or chronic reductions in intake that may be attributed to stress associated with change of diet. In keeping with our earlier work(Reference Shafat, Murray and Rumsey21), we noted that the introduction of the cafeteria diet resulted in immediate increases in energy intake. Cafeteria feeding also greatly increased fat intake at all stages of the experiment (Fig. 2(C) and (D)) and marginally reduced carbohydrate intake (Fig. 2(E) and (F)). Although protein intake tended to be lower in cafeteria diet-fed animals (Fig. 2(G) and (H)), this effect did not achieve statistical significance at any stage of the experiment.
Table 1 Maternal weight gain
(Mean values with their standard errors, n 4–6)
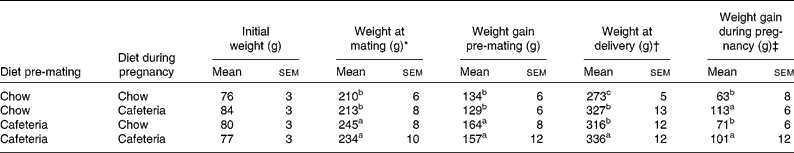
a,b,c Mean values with unlike superscript letters were significantly different (P < 0·05).
* Weight at mating and weight gain from the start of the experiment to mating were influenced by the pre-gestational diet (P = 0·001).
† Weight at the end of pregnancy was influenced by the pre-gestational diet (P = 0·02) and the diet during pregnancy (P = 0·001).
‡ Weight gain during pregnancy was influenced by the diet during pregnancy (P < 0·001).

Fig. 2 Energy and macronutrient intakes of pregnant rats. Values are means for n, as shown in Fig. 1, with their standard errors represented by vertical bars. (A) Energy intake of rats fed the chow diet during lactation. (B) Energy intake of rats fed the cafeteria diet during lactation. Energy intake was significantly influenced by cafeteria feeding during the pre-gestation (*P < 0·001), pregnancy (†P < 0·001) and lactation periods (P < 0·001). (C) Fat intake of rats fed the chow diet during lactation. (D) Fat intake of rats fed the cafeteria diet during lactation. Fat intake was significantly influenced by cafeteria feeding during the pre-gestation (*P < 0·001), pregnancy (†P < 0·001) and lactation periods (P = 0·007). (E) Carbohydrate intake of rats fed the chow diet during lactation. (F) Carbohydrate intake of rats fed the cafeteria diet during lactation. Carbohydrate intake was significantly influenced by cafeteria feeding during the pregnancy (†P = 0·038) and lactation periods (P < 0·001). (G) Protein intake of rats fed the chow diet during lactation. (H) Protein intake of rats fed the cafeteria diet during lactation. –●–, CCC (rats fed the control diet during the pre-gestation, pregnancy and lactation periods); –■–, COC (rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period); –▲–, OCC (rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods); –▾–, OOC (rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period); –○–, CCO (rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period); –□–, COO (rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods); –△–, OCO (rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period); –▽–, OOO (rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods).
The birth weight (Fig. 3(A) and (B)) of female offspring was significantly reduced by exposure to maternal cafeteria feeding pre-gestation (P = 0·010) but only if followed by a chow diet during pregnancy. The cafeteria diet increased the birth weights of male and female rats if fed during pregnancy (P < 0·001) irrespective of the pre-pregnancy diet. Maternal obesity before conception was the main determinant of weight gain from birth to weaning. By weaning at 3 weeks of age, offspring of dams fed the cafeteria diet before mating weighed less than the CCC controls (P = 0·002; Fig. 3(C) and (D)). This effect of the pre-gestational cafeteria diet was greatest if associated with a switch to the chow diet during either pregnancy or lactation. From the age of weaning, male and female animals from the control (CCC) group gained significantly more weight if fed the cafeteria diet post-weaning than if fed a chow diet (Fig. 4), with differences in weight emerging at about 9 weeks of age (after 5 weeks of cafeteria feeding). The other groups showed a similar trend, but weight differences were either less pronounced or appeared later post-weaning.

Fig. 3 Weight of offspring at birth and weaning. (A) Weight of male offspring at birth. (B) Weight of female offspring at birth. (C) Weight of male offspring at weaning. (D) Weight of female offspring at weaning. Weight at birth (n 19–30) was significantly influenced by the sex of the animal (P = 0·014), pre-gestational diet (P = 0·036), diet during pregnancy (P < 0·001) and the interaction of pre-gestational and pregnancy diets (P = 0·012). Weight at weaning (n 16–25) was influenced by pre-gestational diet (P = 0·002), the interaction of pre-gestational and pregnancy diets (P = 0·002) and the interaction of pregnancy and lactation diets (P < 0·001). Values are means, with standard errors represented by vertical bars. a,b,c Mean values with unlike letters were significantly different (P < 0·05). C, cafeteria diet; O, control diet; CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.

Fig. 4 Postnatal growth curves. (A) Weight of groups CCC (rats fed the control diet during the pre-gestation, pregnancy and lactation periods; ○, ●) and COC (rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; □, ■). Curves are shown for male and female offspring (n 3–6). Closed symbols show rats fed the chow diet from weaning. Open symbols show rats fed the cafeteria diet from weaning. (B) Weight of groups CCO (rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; ○, ●) and COO (rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; □, ■). Curves are shown for male and female offspring (n 3–5). Closed symbols show rats fed the chow diet from weaning. Open symbols show rats fed the cafeteria diet from weaning. (C) Weight of groups OCC (rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; ○, ●) and OOC (rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; □, ■). Curves are shown for male and female offspring (n 3–5). Closed symbols show rats fed the chow diet from weaning. Open symbols show rats fed the cafeteria diet from weaning. (D) Weight of groups OCO (rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; ○, ●) and OOO (rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods; □, ■). Curves are shown for male and female offspring (n 3–5). Closed symbols show rats fed the chow diet from weaning. Open symbols show rats fed the cafeteria diet from weaning. Repeated-measures ANOVA indicated that weight was influenced by age (P < 0·001) and interactions of age, with cafeteria diet exposures pre-gestation (P < 0·001), during lactation (P < 0·001), post-weaning (P = 0·039) and with combinations of these factors (age × pre-gestation × lactation, P < 0·001; age × pre-gestation × post-weaning, P = 0·007). Values are means, with standard errors represented by vertical bars.
Offspring weaned on a chow diet
Food intake of all offspring was measured throughout the post-weaning period. For clarity, presentation of data has been limited to the first and fifth weeks post-weaning. As shown in Fig. 5, the early dietary exposures of animals weaned on a chow diet had little impact on energy intake at these time points. By 8 weeks of age (Fig. 5(C) and (D)), male animals consumed more energy than female animals by virtue of their greater body size. The early dietary exposure had little impact on liver weight at 13 weeks of age (Table 2), although male, but not female, animals had smaller livers if exposed to maternal obesity (sex × pre-gestational diet interaction, P = 0·018). Fetal exposure to the cafeteria diet or maternal obesity was associated with reduced adiposity in adulthood. The cafeteria diet pre-mating reduced gonadal fat pad size (P = 0·013) and perirenal fat pad size was similarly reduced in male, but not in female, offspring (sex × pregnancy diet interaction, P = 0·006). Plasma TAG concentrations were also lower in animals exposed to maternal cafeteria feeding in the pre-mating period (P = 0·049). Total cholesterol concentrations were influenced by sex and the maternal diet at all stages of the experiment (four-way interaction of sex × pre-gestational diet × pregnancy diet × lactation diet, P = 0·005), but the main effect was related to cafeteria feeding in lactation, which tended to increase total cholesterol. However, the effects were subtle and there was no strong evidence of hypercholesterolaemia in any of the groups.

Fig. 5 Energy intake of offspring weaned on a chow diet. (A) Energy intake of males in the first week post-weaning (n 4–6). Energy intake was unaffected by early-life exposure to cafeteria feeding. (B) Energy intake of females in the first week post-weaning (n 4–6). Energy intake was unaffected by early-life exposure to cafeteria feeding. (C) Energy intake of males in the fifth week post-weaning (n 4–6). (D) Energy intake of females in the fifth week post-weaning (n 4–6). Energy intake in the fifth week was greater in males than in females (P < 0·001) and was influenced by the interaction of sex, pre-gestational diet and pregnancy diet (P = 0·011). Values are means, with standard errors represented by vertical bars. CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.
Table 2 Body composition and circulating lipids in offspring weaned on a chow diet
(Mean values with their standard errors, n 4–6)

C, chow control diet; O, cafeteria diet; CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.
a,b Mean values with unlike superscript letters were significantly different (P < 0·05).
* Liver and fat pad weights were normalised to body weight after an 18 h fast. Liver size was significantly influenced by sex (P = 0·005) and the interaction of sex and pre-gestational diet (P = 0·018).
† Gonadal fat pad size was influenced by pre-gestational diet (P = 0·013) and the interaction of sex and pregnancy diet (P = 0·006).
‡ Perirenal fat pad size was influenced by sex (P < 0·001), pre-gestational diet (P < 0·001) and the interaction of sex and pre-gestational diet (P = 0·042).
§ Total cholesterol was influenced by pre-gestational diet (P = 0·036), diet during lactation (P = 0·021) and the interaction of sex × pre-gestational diet × pregnancy diet × lactation diet (P = 0·005).
∥ Plasma TAG concentration was influenced by sex (P < 0·001) and the pre-gestational diet (P = 0·049).
Fasting glucose concentrations (baseline blood sample in the glucose tolerance test) of rats weaned on a chow diet were not grossly influenced by the maternal diet during pregnancy. Rats of the OOC group had significantly lower fasting glucose concentrations than those of the OOO group, which was the group with the highest fasting glucose concentrations (pre-gestation × lactation interaction, P = 0·015; Fig. 6). Fasting insulin concentrations varied considerably between the groups, with the females of the COO and OOC groups having markedly lower concentrations than all the other groups of animals (Table 3). As indicated by the smaller AUC (Table 3), female rats cleared glucose more efficiently than male rats. Among the male rats, exposure to the cafeteria diet during the lactation period made glucose clearance more rapid (Fig. 6(A) and (B)). By contrast, it was a chow diet in lactation that was associated with faster glucose clearance in females (OCC and OOC groups; Fig. 6(C) and (D)). There was no evidence of impaired glucose tolerance due to maternal cafeteria feeding in any of the offspring weaned on a chow diet. The insulin responses to the glucose load were variable and showed some impact of early-life programming. Insulin concentrations were determined at 30 min post-administration of glucose to coincide with the peak in plasma glucose. The response was greater in male than in female animals, and based on the change in insulin concentration between baseline and 30 min (Δinsulin; Table 3); there was evidence of an exaggerated insulin response in the females of the COC and OCO groups. Rats in the OOC group, which had the lowest fasting insulin concentrations, also exhibited the lowest Δinsulin. Given that clearance of the intraperitoneal glucose load was similar to the CCC controls, it could be inferred that the rats in this group had greater insulin sensitivity. In male rats, this assertion was supported by significantly lower HOMA-IR (Table 3).

Fig. 6 Glucose tolerance tests in offspring weaned on a chow diet. (A) Males exposed to the chow diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001). (B) Males exposed to the cafeteria diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001). (C) Females exposed to the chow diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001). (D) Females exposed to the cafeteria diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001). Values are means, with standard errors represented by vertical bars. * Mean values were significantly different between the OOC and CCC groups (P < 0·05). † Mean values were significantly different between the OCC and OOC groups (P < 0·05). Fasting glucose concentrations were influenced by the interaction of pre-gestational diet × lactation diet (P = 0·015). –●–, CCC (rats fed the control diet during the pre-gestation, pregnancy and lactation periods); –■–, COC (rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period); –▲–, OCC (rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods); –▾–, OOC (rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period); –○–, CCO (rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period); –□–, COO (rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods); –△–, OCO (rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period); –▽–, OOO (rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods).
Table 3 Insulin concentrations, glucose clearance and expression of insulin signalling pathways in rats weaned on a chow diet
(Mean values with their standard errors, n 4–6)

ipGTT AUC, intraperitoneal glucose tolerance test area under the curve; HOMA-IR, homeostasis model assessment-insulin resistance; Irs2, insulin receptor substrate 2; C, chow control diet; O, cafeteria diet; CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.
a,b Mean values with unlike superscript letters were significantly different (P < 0·05).
* Sex (P < 0·001) and the interaction of pre-gestational diet × lactation diet (P = 0·013) influenced baseline insulin concentrations.
† The 30 min insulin concentration was influenced by sex (P = 0·003) and the interaction of pre-gestation × pregnancy diet (P = 0·002).
‡ Δinsulin was influenced by the same factors (sex, P = 0·027; pre-gestation × pregnancy, P = 0·001). It is the difference between baseline and 30 min insulin concentrations.
§ The AUC for glucose was influenced by sex (P < 0·001) and the interaction of sex × lactation (P = 0·019).
∥ HOMA-IR was influenced by sex (P = 0·001), the interaction of pre-gestation × lactation diet (P = 0·007) and pre-gestation × pregnancy × lactation diet (P = 0·035).
¶ Expression of Irs2 was influenced by sex (P = 0·016) and the interaction of pre-gestation × lactation diet (P = 0·047).
Offspring weaned on a cafeteria diet
As shown in Fig. 7, animals weaned on the cafeteria diet showed little variation in appetite as a consequence of early-life exposures. In the first week post-weaning, rats exposed to the cafeteria diet during suckling consumed less energy (P = 0·023) than those exposed to the chow diet during this period (Fig. 7(A) and (B)), but by 8 weeks of age, this effect had disappeared. All groups of animals weaned on the cafeteria diet consumed more energy (P < 0·001) than those weaned on the chow diet (Fig. 5). As with the animals weaned on the chow diet, there was little effect of the early dietary exposure on liver size (Table 4), although female rats exposed to maternal obesity had larger livers (interaction of sex × pre-gestational diet, P = 0·015). The effects of early dietary exposure on adiposity, which were observed in chow diet-weaned rats, were largely absent in cafeteria diet-weaned offspring, which were all markedly fatter than the chow diet-fed groups (Table 2). Among the cafeteria diet-weaned groups, it was apparent that exposure to the cafeteria diet during lactation significantly increased the size of the perirenal fat pad (P = 0·032), but the size of the effect was minor. Plasma TAG, but not total cholesterol, concentrations (Table 4) were higher (P < 0·001) in animals weaned on the cafeteria diet than those weaned on the chow diet (Table 2). There were no significant effects of maternal diet at any stage on the plasma lipids of cafeteria diet-fed offspring.

Fig. 7 Energy intake of offspring weaned on a cafeteria diet. (A) Energy intake of males in the first week post-weaning (n 4–6). (B) Energy intake of females in the first week post-weaning (n 4–6). Energy intake in the first week post-weaning was influenced by exposure to the cafeteria diet during lactation (P = 0·023). (C) Energy intake of males in the fifth week post-weaning (n 4–6). (D) Energy intake of females in the fifth week post-weaning (n 4–6). Energy intake in the fifth week was greater in males than in females (P < 0·001). Values are means, with standard errors represented by vertical bars. a,b Mean values with unlike letters were significantly different (P < 0·05). CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.
Table 4 Body composition and circulating lipids in offspring weaned on a cafeteria diet
(Mean values with their standard errors, n 4–6)

C, chow control diet; O, cafeteria diet; CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.
a,b Mean values with unlike superscript letters were significantly different (P < 0·05).
* Liver and fat pad weights were normalised to body weight after an 18 h fast. Liver size was significantly influenced by sex (P < 0·001) and the interaction of sex and pre-gestational diet (P = 0·015).
† Gonadal fat pad size was influenced by sex (P < 0·001).
‡ Perirenal fat pad size was influenced by sex (P < 0·001) and the lactation diet (P = 0·032).
As shown in Table 5, fasting insulin concentrations observed in animals weaned on the cafeteria diet were significantly higher (P < 0·001) than those observed in their chow diet-fed littermates (Table 3). As observed in chow diet-fed animals, insulin concentrations were highly variable and were influenced by dietary exposures during pregnancy and lactation. Male rats from the COC, OOC and OOO groups had lower fasting insulin concentrations than the other groups, although variability in the CCC controls reduced the statistical significance of this observation. As shown in Fig. 8, rats weaned on the cafeteria diet developed greater peak glucose concentrations than those weaned on the chow diet (Fig. 6). Considering the AUC for glucose (Table 5), there was clear evidence of glucose intolerance in male rats (COC, COO, OCC and OCO groups) and among some groups of female rats (OCC and OOO) in comparison with the CCC group. Insulin concentrations at 30 min were influenced by the maternal diet during pregnancy, with cafeteria feeding generally resulting in lower plasma insulin. Δinsulin was influenced by sex (P = 0·005), pre-pregnancy diet (P < 0·05) and diet during lactation (P < 0·05). Female rats generally mounted a greater insulin response than male rats. Exposure to maternal obesity (cafeteria diet pre-pregnancy) or the cafeteria diet during pregnancy enhanced the insulin response of the animals, while exposure to the cafeteria diet during lactation had the opposite effect (Table 5). Thus, male rats in the COC, COO, OCC and OCO groups were glucose intolerant, with a strong insulin response to the glucose load. Although this is suggestive of insulin resistance, there was no confirmation of this on the basis of HOMA-IR, which was unchanged by maternal cafeteria feeding at any stage of the experiment. Although the female rats of the OOO group had low baseline insulin concentrations, they mounted a strong insulin response to the glucose load but tended (not statistically significant) to be glucose intolerant.
Table 5 Insulin concentrations, glucose clearance and expression of insulin signalling pathways in rats weaned on a cafeteria diet
(Mean values with their standard errors, n 4–6)

ipGTT AUC, intraperitoneal glucose tolerance test area under the curve; HOMA-IR, homeostasis model assessment-insulin resistance; Irs2, insulin receptor substrate 2; C, chow control diet; O, cafeteria diet; CCC, rats fed the control diet during the pre-gestation, pregnancy and lactation periods; CCO, rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period; COC, rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period; COO, rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods; OCC, rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods; OCO, rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period; OOC, rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period; OOO, rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods.
a,b,c Mean values with unlike superscript letters were significantly different (P < 0·05).
* Sex (P < 0·001), diet in pregnancy (P = 0·026) and the interaction of pregnancy × lactation diet (P = 0·037) influenced baseline insulin concentrations.
† The 30 min insulin was influenced by the pregnancy diet (P = 0·031).
‡ Δinsulin was influenced by sex (P = 0·005), pre-pregnancy diet (P = 0·025) and diet in lactation (P = 0·016). It is the difference between baseline and 30 min insulin concentrations.
§ The AUC for glucose was influenced by the interaction of sex × pre-gestational diet × pregnancy diet (P = 0·024).
∥ HOMA-IR was influenced by sex (P < 0·001).
¶ Expression of Akt2 was influenced by the interaction of pre-gestation, pregnancy and lactation diets (P = 0·004).

Fig. 8 Glucose tolerance tests in offspring weaned on a cafeteria diet. (A) Males exposed to a chow diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001). (B) Males exposed to the cafeteria diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001) and by the interaction of time × pre-gestational diet × pregnancy diet (P = 0·022). (C) Females exposed to the chow diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001). (D) Females exposed to the cafeteria diet during lactation (n 4–6). Glucose concentration was influenced by time post-intraperitoneal administration of glucose (P < 0·001) and by the interaction of time × pre-gestational diet × pregnancy diet (P = 0·018). Values are means, with standard errors represented by vertical bars. * Mean values were significantly different between the COC and CCC groups (P < 0·05). † Mean values were significantly different between the OCC and CCC groups (P < 0·05). ‡ Mean values were significantly different between the OCO and CCO groups (P < 0·05). § Mean values were significantly different between the COO and OOO groups (P < 0·05). –●–, CCC (rats fed the control diet during the pre-gestation, pregnancy and lactation periods); –■–, COC (rats fed the control diet during the pre-gestation period, the cafeteria diet during the pregnancy period and the control diet during the lactation period); –▲–, OCC (rats fed the cafeteria diet during the pre-gestation period and the control diet during the pregnancy and lactation periods); –▾–, OOC (rats fed the cafeteria diet during the pre-gestation and pregnancy periods and the control diet during the lactation period); –○–, CCO (rats fed the control diet during the pre-gestation and pregnancy periods and the cafeteria diet during the lactation period); –□–, COO (rats fed the control diet during the pre-gestation period and the cafeteria diet during the pregnancy and lactation periods); –△–, OCO (rats fed the cafeteria diet during the pre-gestation period, the control diet during the pregnancy period and the cafeteria diet during the lactation period); –▽–, OOO (rats fed the cafeteria diet during the pre-gestation, pregnancy and lactation periods).
To investigate the basis of insulin resistance in animals exposed to a cafeteria diet in early life, we examined the mRNA expression of the two components of the insulin signalling pathway in the liver. Among the rats weaned on a chow diet, there was little effect of maternal diet upon the expression of Irs2 or Akt2, although among male rats, maternal obesity lowered Irs2 expression, but only if coupled with a chow diet during lactation (Table 3). Among the animals weaned on a cafeteria diet (Table 5), there was an up-regulation of both Irs2 and Akt2 expressions in the male rats of the OCC group, relative to the CCC controls.
Discussion
The aim of the present study was to determine whether early-life exposure to maternal cafeteria feeding had an impact on glucose homeostasis in young adult rats. The basis for the experiment was the observation that mice exposed to maternal high-fat feeding were programmed to develop glucose intolerance, in a manner analogous to epidemiological evidence suggesting that the maternal diet in early development may exert a programming influence on metabolic function(Reference Eriksson, Forsén and Tuomilehto3, Reference Samuelsson, Matthews and Argenton18, Reference Pirkola, Pouta and Bloigu26). The present experiment had a complex design, but this was necessary in order to fully model whether or not maternal obesity, or simply overfeeding on a fat- and sugar-rich diet, provided the programming stimulus leading to a compromised metabolic phenotype. Although primarily observational, the present study has identified the separate contributions of obesity and overnutrition, at different developmental stages, to programming glucose homeostasis and will be a valuable stimulus for more focused, mechanistic, follow-up studies.
An earlier study(Reference Akyol, Langley-Evans and McMullen20) has established that the cafeteria diet protocol induced obesity in young female rats, with no adverse impact on their reproductive capacity. In the present study, we again observed that feeding a cafeteria diet for 7 weeks before mating resulted in excess weight gain, and although body fat measurements were not possible, the earlier work(Reference Akyol, Langley-Evans and McMullen20) enables us to conclude that the feeding protocol rendered the young adult female rats obese. As a result, considering the offspring of the OCC and OCO groups allowed us to model the impact of maternal overweight during pregnancy independently of any influence of overfeeding during pregnancy. Introducing the cafeteria diet from the start of pregnancy (COC and COO groups) allowed consideration of the impact of overfeeding during fetal development in the absence of maternal obesity before conception, while the other four cafeteria diet-fed groups enabled consideration of the interactive effects of maternal overweight and overfeeding.
Exposure to the maternal cafeteria diet at any developmental stage before weaning had no impact on body weight, food intake, adiposity or circulating lipids in the offspring. While some studies have suggested that maternal overfeeding drives weight gain and hyperphagia(Reference Shankar, Harrell and Liu17–Reference Bayol, Simbi and Bertrand19), the present findings are consistent with a robust literature suggesting that exposure to greater concentrations of maternal leptin, particularly in milk, results in resistance to overweight in animals of equivalent age(Reference Picó, Oliver and Sánchez27) to those in the present study and older(Reference Sánchez, Priego and Palou28). While we did not measure leptin in milk, increased leptin secretion may be a reasonable assumption in rat dams rendered obese by cafeteria feeding before and during pregnancy. The overall quality of milk produced by cafeteria diet-fed dams would be an interesting area for further investigation, as this may help explain the outcomes in the offspring. Rats fed high-fat diets have been shown to produce milk that has a high content of protein and lactose as well as lipids(Reference Del Prado, Delgado and Villalpando29). The impact of this specific exposure on offspring development is yet to be evaluated. While overnutrition induced by reduction in litter size is known to induce hyperphagia and obesity in young rats(Reference Gorski, Dunn-Meynell and Hartman30, Reference Plagemann, Harder and Rake31), pup overnutrition through excessive milk consumption is not necessarily equivalent to greater macronutrient density in milk.
When weaned on a cafeteria diet, all groups of animals had larger fat depots and higher circulating TAG, and were heavier than those weaned on a chow diet, but there were no differences related to maternal diet at any stage of development. These findings are in stark contrast with those of Bayol et al. (Reference Bayol, Simbi and Bertrand19) who employed an almost identical protocol but observed profound hyperphagia and greater adiposity following maternal cafeteria feeding. These discrepancies are difficult to explain but may relate to the Bayol study(Reference Bayol, Simbi and Bertrand19), not using a pre-mating run-in to the introduction of cafeteria feeding and employing a protocol in which the variety in the cafeteria diet was greater than that in the present study.
The main finding of the present study was that glucose intolerance, as evidenced by the greater AUC for glucose, could be programmed, independently of any other gross metabolic disturbance, by exposure to maternal cafeteria feeding before and during pregnancy, and during lactation. This is in broad agreement with other studies(Reference Shankar, Harrell and Liu17, Reference Samuelsson, Matthews and Argenton18, Reference Bayol, Simbi and Fowkes32–Reference Shankar, Kang and Harrell34), although in most cases, glucose intolerance has been shown to be part of a broader spectrum of metabolic disturbances. In rats, abnormal glucose homeostasis can be programmed along with obesity by overfeeding mothers with high-fat–high-sugar diets(Reference Nivoit, Morens and Van Assche33) and intra-gastric feeding with an energy-dense liquid feed(Reference Shankar, Harrell and Liu17, Reference Shankar, Kang and Harrell34). However, in the present study, the appearance of the glucose-intolerant phenotype by 3 months of age was wholly dependent upon post-weaning exposure to an energy-dense, obesity-inducing cafeteria diet. When weaned on a low-fat chow diet, there was little evidence of metabolic disturbance, although there was evidence of greater insulin sensitivity among some groups of offspring exposed to maternal obesity in utero. Similar observations have been made in the offspring of rats fed a low-protein diet during pregnancy, where the apparent early sensitivity to insulin observed in younger offspring gives way to insulin resistance in older adulthood(Reference Erhuma, Salter and Sculley35, Reference Langley, Browne and Jackson36).
It was intended that the design of the experiment would give an insight into the critical period of development that may influence later development of metabolic disease and identify whether maternal obesity or maternal overfeeding was the main driving force for these effects. The differences between the present study and work in mice using high-fat–high-sugar diets during pregnancy(Reference Samuelsson, Matthews and Argenton18, Reference Shankar, Kang and Harrell34) may suggest that overfeeding during pregnancy is a key factor in the programming of offspring adiposity and dyslipidaemia, but that overweight is the driver of glucose intolerance. Although we found that cafeteria feeding at all stages of the present study had an influence on glucose homeostasis, the most consistent observation was that the cafeteria diet pre-mating, i.e. the condition of maternal overweight, was associated with offspring glucose intolerance. The cafeteria diet during pregnancy had only minor independent effects on glucose homeostasis and tended to lower baseline insulin. It is possible therefore that overfeeding may have an impact on pancreatic development, while obesity may exert effects on insulin signalling pathways. The present data indicated that the development of glucose intolerance was possibly at an early stage. While the data on the AUC indicated intolerance, it was clear that differences were largely a product of greater peak glucose concentrations, with no marked impairment of clearance at 2 h post-glucose injection. Similarly, HOMA-IR showed no evidence of insulin resistance even though data on baseline insulin concentrations, Δinsulin and AUC for glucose were often suggestive of an insulin-resistant state. It is possible that with ageing, glucose intolerance may become more pronounced and more clearly associated with impaired insulin signalling. Age-dependent emergence of an insulin-resistant phenotype has been previously reported in the context of programming by maternal undernutrition(Reference Erhuma, Salter and Sculley35).
Among the studies that have considered the impact of early-life exposure to maternal overnutrition on glucose homeostasis, there is considerable variation in the exact nature of the programmed phenotype and the mechanisms that underpin the development of glucose intolerance. Low insulin concentrations observed in some groups in the present study suggest that similar to the offspring of mice fed high-fat diets(Reference Samuelsson, Matthews and Argenton18), pancreatic β-cell exhaustion may have contributed to impaired glucose homeostasis. Other studies have suggested the programming effects of maternal overfeeding on insulin signalling and insulin-responsive lipogenic pathways(Reference Samuelsson, Matthews and Argenton18, Reference Nivoit, Morens and Van Assche33, Reference Shankar, Kang and Harrell34). The present study has some commonality with such studies as we observed up-regulation of Akt2 in insulin-resistant male rats weaned on a cafeteria diet. Although Irs2 and Akt2 expression was increased in the insulin-resistant OCC male group, there is limited evidence that insulin signalling pathways were grossly perturbed in a direction that favoured insulin resistance. Our study could not exclude the possibilities that the cafeteria diet programmed the expression of the insulin receptor itself, or other components of the signalling pathway, such as phosphatidylinositol 3 kinase, or that programmed effects were mediated at the level of protein rather than RNA expression. The liver is, of course, just one tissue involved in the uptake and disposal of glucose, and the present study did not include possible contributions of the insulin signalling pathway to the skeletal muscle or the adipose tissue, which have been previously shown to be sensitive to maternal overfeeding(Reference Bayol, Simbi and Fowkes32, Reference Bayol, Simbi and Stickland37). These issues will have a high priority for resolution in further studies. Mechanistically, it is difficult to explain the pattern of results observed in the offspring. In terms of the signal from mother to fetus, which initiates the metabolic adaptations that subsequently manifest as glucose intolerance, it may be that the obesity induced by cafeteria feeding exacerbates the normal insulin resistance of pregnancy and increases the metabolic load on the developing liver and pancreas. Alternatively, maternal obesity could interfere with the normal maternal–fetal endocrine balance across the placenta. These are issues that should be explored in future investigations.
In common with a number of other studies, we observed sex differences in the programmed response to the maternal cafeteria diet. Male offspring were more susceptible to the programming of glucose homeostasis than female offspring. Bayol et al. (Reference Bayol, Simbi and Fowkes32) reported that there were differences in the hepatic glucose uptake between male and female offspring following maternal cafeteria feeding. Nivoit et al. (Reference Nivoit, Morens and Van Assche33) also reported that male offspring were more susceptible to obesity, hyperphagia and insulin resistance than female offspring, following exposure to a high-fat–high-sugar maternal diet during fetal development and suckling. Sex differences in the programming of glucose homeostasis have been reported in the offspring of rats fed low-protein diets during pregnancy and lactation, with male rats developing an insulin-resistant, hyperinsulinaemic state as young adults(Reference Sugden and Holness38). The mechanistic basis of these differences is yet to be explained. Fernandez-Twinn et al. (Reference Fernandez-Twinn, Wayman and Ekizoglou39) reported that while female offspring from protein-restricted mothers developed insulin resistance considerably later than male offspring, the basic underlying mechanisms were identical, suggesting that the sexes differ only in their profiles of age-related disease. In contrast, Chamson-Reig et al. (Reference Chamson-Reig, Thyssen and Hill40) suggested that in female rats, pancreatic insufficiency may drive impaired glucose homeostasis following maternal protein restriction, with insulin signalling defects being confined to male rats.
The present study has demonstrated the programming of glucose intolerance in the offspring of obese and overnourished mothers, independently of any effect on offspring adiposity. The present data suggest that maternal obesity before pregnancy is sufficient to programme glucose homeostasis in the resulting offspring. The mechanisms that lead to dysfunction of glucose homeostasis may vary according to the timing of additional maternal overfeeding insults. Further research will be required to fully define these mechanisms, the processes that link from maternal metabolic status to function in the offspring and the nature of the differences in response to maternal obesity between male and female animals.
Acknowledgements
The expert technical support of Thom Wright, Carol Armett, Sarah Kirkland and Richard Plant is gratefully acknowledged. A. A. was funded by a studentship from Hacettepe University (Ankara, Turkey). The authors declare that they have no conflicts of interest. The authors' contributions were as follows: S. C. L.-E. and S. M. designed the experiment. A. A. performed the experimental and laboratory analyses, and collated the data. S. C. L.-E. and A. A. performed the statistical analyses. S. C. L.-E. wrote the manuscript. All authors read and approved the final content.













