

INTRODUCTION
Influenza A is a highly diverse virus and is therefore classified according to the two glycoproteins that are presented on the virus membrane, the haemagglutinin (HA) and neuraminidase (NA). Since infected poultry may develop a wide variety of symptoms, avian influenza viruses (AIVs) are additionally classified according to their virulence in poultry [Reference Alexander1, Reference Webster2]. Highly pathogenic avian influenza viruses (HPAIVs) cause severe sickness and high mortality rates in affected poultry holdings. Outbreaks of these viruses may lead to serious epidemics with disastrous consequences that can affect a large region [Reference Alexander1]. To date, HPAIVs have mainly been found in H5 and H7 subtypes. AIVs that cause no or minor symptoms in poultry are classified as low-pathogenic avian influenza viruses (LPAIVs). However, infections with LPAIV may result in more severe symptoms if they are combined with poor air quality in poultry barns or secondary infections [Reference Halvorson3]. LPAIVs can be found in HA subtypes 1–16 [Reference Fouchier4]. Whereas LPAIV outbreaks are usually of limited economic importance, a spontaneous insertion of basic amino acids at the HA0 cleavage site may give rise to a highly pathogenic variant of the strain [Reference Alexander1]. This was witnessed in some past H5 and H7 LPAIV outbreaks in Pennsylvania (1983), Mexico (1994–1995) and northern Italy (1999–2000) [Reference Capua5–Reference Horimoto7].
AIVs can be introduced in commercial poultry holdings or smallholder flocks in numerous ways. Human activity or movements from other mechanical vectors such as rodents, passerines or other animals may cause such passive introduction without infection [Reference Olsen8]. Direct contact with pigs or humans infected with avian influenza (AI) has been observed as a possible route for introduction of AI as well [Reference Mohan9, Reference Wood10]. However, direct contact with infected waterfowl is considered to be the most important route for the introduction of LPAI to poultry [Reference Alexander1]. Indeed, for most HPAI outbreaks in poultry that have been reported during the past 5 decades, it has been shown that a wild bird LPAIV had been introduced to the index case and then mutated into a HPAIV [Reference Koch and Elbers11]. Since the beginning of AI surveillance studies in the early 1970s, a large number of LPAI viruses have been isolated in different wild bird species [Reference Alexander and Brown12, Reference Lupiani and Reddy13]. Of these, wild aquatic birds, particularly anseriformes and charadriiformes, are now known to be the natural reservoir of LPAIV [Reference Olsen8]. Therefore, the possibility of direct or indirect contact between poultry and wild birds is considered a major risk for the introduction of the virus and the subsequent emergence of a HPAIV, if the virus is allowed to spread [Reference Koch and Elbers11, Reference Alexander and Brown12]. However, this natural route of LPAIV introduction to poultry has never been observed closely, and important information on this occurrence is lacking. Therefore, studying the transmission of LPAIVs from wild birds to poultry in experimental conditions may provide more insight into the dynamics of this event and on the exact route by which the virus is transmitted.
In this study, a wild bird-originated H5N3 LPAIV was at first phenotypically characterized in specific pathogen free (SPF) chickens and Pekin ducks. Then, the introduction of the virus from infective Pekin ducks to susceptible SPF chickens was studied in a series of transmission experiments targeting direct virus transmission through (i) close contact between infected ducks and chickens and (ii) contaminative transmission.
MATERIALS AND METHODS
Virus
All experiments were conducted with LPAIV H5N3 A/Anas platyrhynchos/09-884/2008. The virus was isolated from a cloacal swab of a mallard duck that was sampled in La Hulpe (Belgium) in December 2008 as part of the Belgian long-term wild-bird monitoring programme [Reference Van14]. The swab was found positive by one-step real-time reverse transcriptase–polymerase chain reaction (rRT–PCR), and the virus was isolated in 9-day-old embryonated SPF chicken eggs. For this study, a second passage of the virus was diluted in sterile phosphate-buffered saline to obtain an inoculum of 107 egg infectious dose (EID)50/ml.
Animals
One-day-old white Pekin ducks (Anas platyrhynchos) were purchased from a local producer (Wijverkens pluimvee, Belgium). SPF chicken eggs were purchased from Lohman-Valo (Germany) and hatched in our facilities, under biosafety level 3 (BSL-3) conditions. All animals were housed in BSL-3 isolators (type: HM1500, Montair Process Technology B.V., The Netherlands) until the end of the experiment. The inner floor surface of the isolators measured 80 × 150 cm and the height was 72 cm. The isolator floor comprised of a stainless-steel grid covered with a plastic grid to allow more grip for the animals. In the transmission experiments, half of the grid area was covered with plastic, to prevent faeces from passing through. The walls and ceiling of the isolator are stainless-steel and two walls contain acrylic glass with rubber gloves allowing manipulation of the animals. The animals received feed from a stainless-steel feeder and tap water from a plastic automated bell drinker, unless stated otherwise. A negative air pressure of 45 ± 5 m3/h was maintained during the entire course of the experiment. Each animal experiment was conducted under the authorization and supervision of the Biosafety and Bioethics Committee at the Veterinary and Agrochemical Research Centre, following national and European regulations.
Experimental design
SPF chicken infection experiment (experiment 1)
Twelve SPF chickens were oculo-nasally inoculated with 100 μl of the inoculum. Virus shedding was then followed by collecting oropharyngeal (OP) and cloacal (CL) swabs at 1, 3, 6 and 10 days post-inoculation (dpi). Immune response was assessed by collecting blood samples at 7, 10, 14 and 21 dpi.
Transmission experiments involving close contact (experiment 2)
This experiment consisted of three trials (2a, 2b, 2c). Trials 2a and 2b each started with a group of six Pekin ducks which were oculo-nasally inoculated with 100 μl of the inoculum and then placed in an isolator. Trial 2c started with a group of three Pekin ducks which were inoculated and placed in a third isolator. The day of inoculation is hereafter referred to as day −1. In all trials, OP and CL swabs were collected from the inoculated Pekin ducks at day 0, after which each group of ducks was transferred to an isolator where six susceptible SPF chickens were housed. Then, OP and CL swabs were collected from every duck and chicken at days 1, 2, 3, 4, 6, 10, 14, 18 and 21. At the same time that the animals were sampled, floor swabs and drinking-water samples were collected to assess the environmental infection pressure. Blood samples from all animals were collected at 14 and 21 dpi and 14 and 21 days post-exposure (dpe) (corresponding to days 13 and 20 for Pekin ducks and days 14 and 21 for SPF chickens). In the Results section this experiment is presented in two parts: the assessment of the infectivity of H5N3 for Pekin ducks (experiment 2, part 1) and the transmission experiment between ducks and chickens involving close contact (experiment 2, part 2).
Transmission experiments involving contaminative transmission (experiment 3)
This experiment consisted of two trials (3a, 3b), which were replicates. Each trial consisted of two groups of six susceptible SPF chickens, the drinking-water exposure (DWE) group and the surface exposure (SE) group, the names referring to the fomite to which these chickens were exposed. The trials were designed as follows (Fig. 1): three Pekin ducks were inoculated at day −4 and housed in an isolator for 4 days, until day 0. Instead of using the automated bell drinker that was used in all other experiments, the ducks received drinking water in a 2·5-l polypropylene drinking bowl that was replenished daily. At day 0, also referred to as the replacement day, the ducks were removed from the isolator; the drinking bowl was placed in the isolator housing the DWE group and the chickens from the SE group were placed in the isolator where the ducks were previously housed. Chickens of the DWE group were thus exposed to contaminated drinking water and chickens of the SE group were thus exposed to (primarily faecally) contaminated dry surfaces such as isolator walls, floor, feeder and feed. An automated bell drinker was installed in the SE group to provide non-contaminated drinking water. Since contamination was induced by the same three inoculated Pekin ducks for both the DWE and SE groups, a comparison of the importance of these fomites for LPAIV transmission was possible. OP and CL swabs were collected from all susceptible SPF chickens at days 1–5, 7, 10 and 14. The environmental infection pressure was assessed by taking floor swabs and drinking-water samples at days 1–5, 7, 10, 12 and 14. Blood samples were collected from all chickens at days 14 and 21.
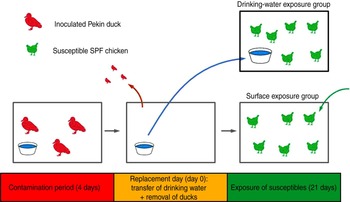
Fig. 1 [colour online]. Experimental design of direct transmission experiments involving contaminative transmission. Three inoculated Pekin ducks were housed in an isolator for 4 days. At the end of this 4-day period, the ducks were removed from the isolator. Then, the drinking bowl was placed in a different isolator where six susceptible specific pathogen free (SPF) chickens were housed (drinking-water exposure group); and six other susceptible SPF chickens were placed in the isolator where the ducks were previously housed (surface exposure group). Virus transmission to SPF chickens from both groups was then monitored during a period of 21 days.
Sample handling
Animal and floor swabs were immediately immersed in 1·5 ml of a storage medium [brain-heart-infusion (BHI) broth enriched with a mixture of antibiotics containing gentamycin, kanamycin, penicillin and streptomycin (BHI+AB)], after which the sample was briefly vortexed to release swab material and the cotton was discarded. Drinking-water samples (1·5 ml) were poured into 1·5 ml of a double-concentrated BHI+AB storage medium, to yield the same concentration of medium and antibiotics in drinking-water samples as that present in swab samples. All samples were stored at −80°C, awaiting further analysis. Blood samples were allowed to coagulate at room temperature after which the serum was harvested and stored at −20°C until testing for antibodies was performed.
Detection and quantification of viral RNA in samples using rRT–PCR
Samples were allowed to reach room temperature and viral RNA (vRNA) was semi-automatically extracted from 50 μl sample material using a KingFisher magnetic particle processor and the MagMax™ AI/ND-96 VRNA kit (Ambion Inc., USA), according to the manufacturer's protocol. A total of 25 μl reaction volume (containing 2 μl of purified RNA) was prepared using the Quantitect Probe RT–PCR kit (Qiagen GmBH, Germany) and amplification of the matrix gene was carried out in a Biosystems 7500 real-time PCR cycler (Applied Biosystems, Belgium) [Reference Van15]. In each run, a series of eight 1:10 dilutions of synthetic matrix RNA was run simultaneously to calculate the number of RNA copies per ml sample medium. Animals were considered positive by rRT–PCR when at least one swab sample taken throughout the course of the experiment contained at least 100 copy of vRNA per reaction volume, thus corresponding to 102·7 vRNA copies/ml sample medium.
Serology
Serum samples were tested for the presence of antibodies directed towards the viral nucleoprotein with the ID Screen influenza A antibody competition ELISA kit (Idvet, France). All tests were conducted according to the manufacturer's instructions. In the data analysis, serum samples with a sample-to-negative (S/N) ratio of ⩾0·5 were considered negative, and samples with a S/N ratio <0·5 were considered positive. Animals were considered positive by serology if at least one serum sample was found positive by nucleoprotein (NP)-ELISA.
Virus titration using chicken embryo fibroblasts (CEFs)
Quantification of AIV in water samples and floor swabs was performed using a microtitre endpoint titration in primary cultures of CEFs. Final cell suspensions of 2 × 105 CEF/ml were prepared in L15/Leibovitz+McCoy medium enriched with gentamycin and glutamine and allowed to adhere to the wells of a 96-well microtitre plate for 2 days in the presence of inactivated fetal bovine serum. A more detailed protocol can be found elsewhere [Reference Stallknecht16]. The samples were diluted in L15/Leibovitz+McCoy medium enriched with gentamycin, glutamine and TPCK trypsin and incubated. The wells were examined for cytopathic effect by light microscopy. Fifty per cent tissue culture infectious dose/ml sample medium (TCID50/ml) was calculated using the method described by Reed & Muench [Reference Reed and Muench17].
Statistical analysis
The transmission of LPAIV H5N3 A/Anas platyrhynchos/09-884/2008 by close contact (experiment 2) was modelled in a stochastic susceptible-infectious (SI) transmission model. We assumed unequal infectivities and susceptibilities for ducks and chickens, and considered our study population to be heterogeneous. Theoretically, four different transmission parameters can thus be considered to play a part in experiment 2; transmission from ducks to chickens (β dc), transmission between chickens (β cc), transmission from chickens to ducks (β cd) and transmission between ducks (β dd), analogous to Velthuis et al. [Reference Velthuis18]. Since all ducks were inoculated at the beginning of the experiment, only β dc and β cc were considered in the statistical analysis. Similarly, in a previous study investigating transmission of the same virus between chickens, β cc could not be determined since transmission did not occur [Reference Claes19]. Therefore, it was assumed that β cc = 0 and that all infected chickens became infected by duck–chicken virus transmission, quantified by β dc. The transmission parameter β dc and its 95% confidence interval (CI) was estimated with a generalized linear model in SPSS v. 19 (SPSS Inc., USA), using a complementary log-log function and the offset function:ln(IdΔt/N), where Id represents the number of infectious ducks and N represents the total number of animals at the beginning of the time interval ∆t. Susceptible chickens were considered infected if anti-AIV antibodies were detected in blood serum at either 14 or 21 dpe. The time of infection was determined as the first day on which an OP swab from an infected chicken was found positive by rRT–PCR.
Comparing overall quantities of vRNA in OP and CL swabs between experiments was performed by calculating median area under the curve (AUC) values [Reference Brown20, Reference Spekreijse21]. Hereto, quantities of vRNA in OP and CL swabs of each bird were plotted over time and the AUC was calculated in Microsoft Excel (Microsoft Corporation, USA), using the following formula:
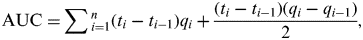 $${\rm AUC =} \sum {_{i = 1}^n (t_i - t_{i - 1} )q_i + \displaystyle{{(t_i - t_{i - 1} )(q_i - q_{i - 1} )} \over 2}}, $$
$${\rm AUC =} \sum {_{i = 1}^n (t_i - t_{i - 1} )q_i + \displaystyle{{(t_i - t_{i - 1} )(q_i - q_{i - 1} )} \over 2}}, $$
where q i is the number of vRNA copies or TCID50/ml storage medium for the sample at time t i . Only animals that were positive by rRT–PCR for at least one swab sample were considered. AUC values were thus calculated for each bird and the median, 25th and 75th percentiles are given in the Results section.
Relationships between vRNA quantities in duck and chicken OP and CL swabs and environmental samples were assessed with an analysis of variance (ANOVA), which was performed in SPSS v. 19. A log10(1+x) transformation was performed.
RESULTS
Experiment 1: infectivity of H5N3 A/Anas platyrhynchos/09–884/2008 for SPF chickens
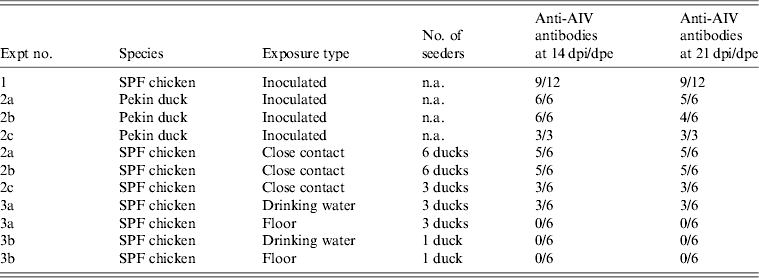
At 1 dpi, vRNA was detected in OP swabs from 5/12 chickens, of which three chickens were still found positive at 3 dpi (Fig. 2 a). At 6 and 10 dpi, vRNA was no longer detected in OP swabs (Fig. 2 a). CL swabs were all negative, except for one swab taken at 3 dpi from a chicken that was also found positive for OP shedding (Fig. 2 b). An overall estimation of vRNA quantities shed by each chicken throughout the entire course of the experiment was made by calculating the AUC of vRNA quantities found in each sample plotted over time. These results show that OP viral shedding in inoculated SPF chickens is the strongest, while CL virus shedding is almost negligible (Table 1). However, seroconversion was observed in more chickens than virus shedding was. Indeed, anti-AIV antibodies were demonstrated in serum samples from 9/12 inoculated SPF chickens at both 14 and 21 dpi (Table 2).
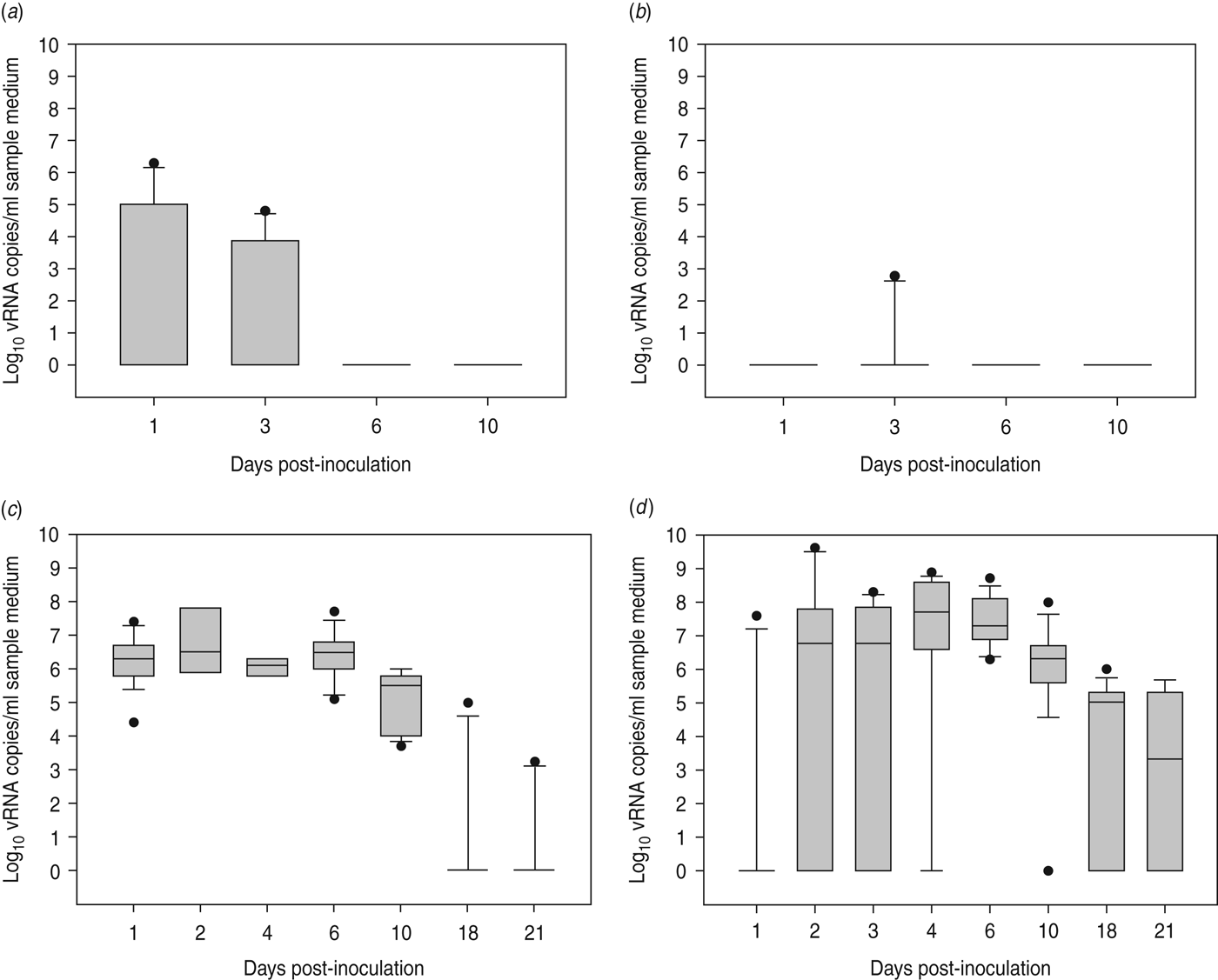
Fig. 2. Virus shedding patterns for specific pathogen free chickens and Pekin ducks inoculated with H5N3 A/Anas platyrhynchos/09–884/2008. Log10 vRNA copies/ml sample medium are presented for (a) oropharyngeal SPF chickens swabs, (b) cloacal SPF chicken swabs, (c) oropharyngeal Pekin duck swabs and (d) cloacal Pekin duck swabs. Negative samples are represented as 100.
Table 1. Overview of the total amount of viral RNA found in oropharyngeal and cloacal swabs from chickens and ducks throughout the entire course of each trial
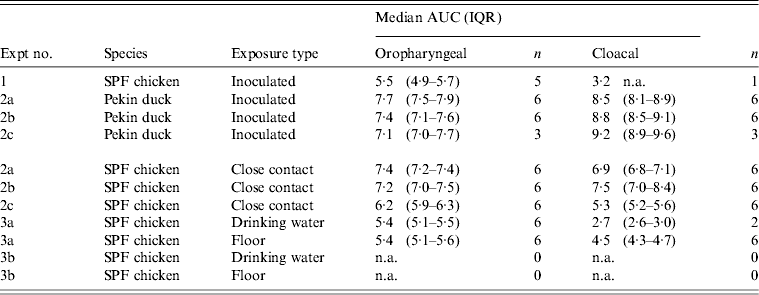
AUC, Area under the curve; IQR, interquartile range; SPF, specific pathogen free; n.a., not available.
Log10 of the median and the IQR of the AUC that were calculated for each animal separately are shown for each trial. n = the number of animals that were positive by rRT–PCR at least once, and on which data for AUC calculations are based.
Table 2. Overview of NP-ELISA serology results for all trials conducted in the present study
AIV, Avian influenza virus; dpi, days post-inoculation; dpe, days post-exposure; n.a., not available; SPF, specific pathogen free.
Experiment 2, part 1: infectivity of H5N3 A/Anas platyrhynchos/09–884/2008 for Pekin ducks
All 15 Pekin ducks (six from trial 2a, six from trial 2b, three from trial 2c) showed OP and CL virus shedding; OP virus shedding started at 1 dpi while the onset of CL virus shedding varied (Fig. 2 c, d). One duck from trial 2c already exhibited CL shedding at 1 dpi, although this commenced at 2 dpi for all ducks from trial 2a and the two remaining ducks from trial 2c. The onset of CL virus shedding was more delayed in trial 2b: 2/6 ducks at 3 dpi, 1/6 ducks at 4 dpi and 2/6 ducks at 6 dpi (samples from 5 dpi were not analysed). Overall estimation of the routes and intensity of virus shedding throughout the infectious period showed a completely different profile for Pekin ducks than for SPF chickens. While virus shedding in SPF chickens is primarily through the oropharynx, CL virus shedding is the strongest virus shedding route in ducks. Moreover, the overall intensity of virus shedding was higher in ducks than in SPF chickens (Table 1). Seroconversion was seen in all inoculated Pekin ducks at 14 dpi. However, the presence of antibodies in this species did not appear as long-lasting since only 12/15 Pekin ducks remained positive by NP-ELISA at 21 dpi (Table 2).
Experiment 2, part 2: transmission experiment between ducks and chickens involving close contact
Virus shedding and seroconversion in inoculated Pekin ducks have been discussed before. In all three trials, all susceptible SPF chickens were positive by rRT–PCR. In fact, all OP and most CL swabs taken from these chickens were positive by rRT–PCR for several consecutive days starting at 1 dpe for trial 2a, 3 dpe for trial 2b and 1 dpe for trial 2c. These time points each coincided with the onset of CL virus shedding that was observed in Pekin ducks (see above). The profile of vRNA detection in OP and CL SPF chicken swabs was different from that observed in experiment 1 (Fig. 3). Not only were more CL swabs found positive within 10 dpe (25/30 for trial 2a; 16/30 for trial 2b, 14/24 for trial 2c), but also the difference in vRNA quantities for the two swab types was smaller (Table 1). Moreover, the amounts of vRNA found in both OP and CL swabs were many times larger than those found in experiment 1, as witnessed by the AUC values shown in Table 1. Fewer SPF chickens were positive by serology than by rRT–PCR (Table 2). Based on the duration, intensity and routes of virus shedding, the chickens that seroconverted were not readily distinguishable from those that did not. In the two trials where six inoculated Pekin ducks were used, anti-AIV antibodies were detected in 5/6 SPF chickens (trials 2a, 2b). In the trial where only three inoculated Pekin ducks were used, anti-AIV antibodies were detected in 3/6 SPF chickens (trial 2c). For the three trials combined, β dc was estimated at 0·30 (95% CI 0·18–0·52).
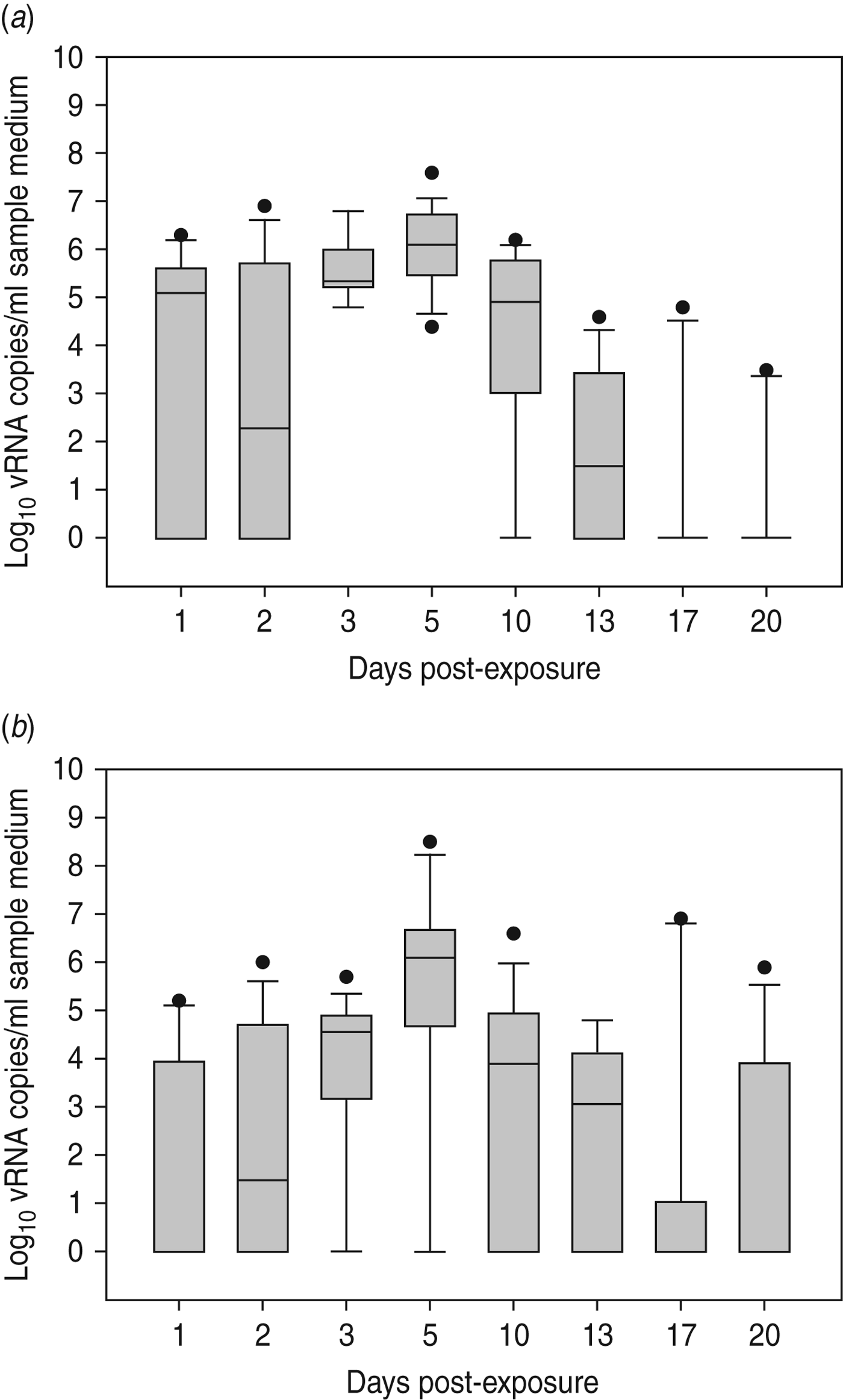
Fig. 3. Presence of viral RNA in oropharyngeal and cloacal swabs from specific pathogen free (SPF) chickens that were exposed by close contact to Pekin ducks inoculated with H5N3 A/Anas platyrhynchos/09–884/2008. Log10 vRNA copies/ml sample medium are presented for (a) oropharyngeal SPF chickens swabs and (b) cloacal SPF chicken swabs. Negative samples are represented as 100.
Large quantities of vRNA were demonstrated in drinking-water samples and floor swabs from all trials. In trials 2a and 2c, vRNA was first detected in these samples at 1 dpe whereas in trial 2b (where duck CL virus shedding was delayed), vRNA was first detected in drinking water at 3 dpe and on the floor of the isolator at 2 dpe. To assess the relationship between vRNA in swabs from inoculated ducks, drinking-water samples and floor swabs, an ANOVA was performed. These results indicated significant associations between vRNA in duck CL swabs for drinking water (P < 0·01) and floor swabs (P < 0·01). However, no significant relationship was found between vRNA in duck OP swabs for drinking water (P = 0·395) and floor swabs (P = 0·197). For SPF chickens, the same analysis indicated a significant relationship between vRNA in CL swabs for drinking water (P < 0·01) and floor swabs (P = 0·023), and also between vRNA in OP swabs for drinking water (P < 0·01) and floor swabs (P < 0·01).
Experiment 3: transmission experiment between ducks and chickens involving contaminative transmission
Trial 3a
In trial 3a, all three inoculated Pekin ducks were successfully infected. At the replacement day, the drinking water was visually contaminated with bird faeces and contained 108·6 vRNA copies/ml drinking water. Similarly, the floor swab of the isolator contained 108·4 vRNA copies/ml storage medium (Table 3).
Table 3. Transmission experiments involving contaminative transmission. Trial 3a: assessment of the environmental contamination. Overview of the number of vRNA copies and 50% tissue culture infectious dose (TCID50) in drinking-water samples (drinking-water exposure group) and floor swabs (surface exposure group).

dpe, Days post-exposure; CEF, chicken embryo fibroblasts; AUC, area under the curve.
Results are expressed as log10 vRNA copies/ml sample medium or log10 TCID50/ml sample medium. An overview of the total amount of vRNA copies and TCID50 in samples obtained between the day of exposure and 7 dpe is given by calculation of the AUC.
+, Positive sample (titre not determined); –, negative sample.
For each susceptible SPF chicken from the DWE group, one or several OP swabs were positive by rRT–PCR. In total, 23/48 OP swabs, analysed between 0 and 14 dpe were found positive by rRT–PCR. By contrast, a total of only 2/48 CL swabs, coming from two different chickens, were found positive during the same time period. Results from AUC calculations show that the quantities of vRNA found in the OP swabs throughout the entire course of the trial were greater than those found in CL swabs, which contained only traces of vRNA (Table 1). The AUC values for both swab types are comparable to what was observed in experiment 1. However, we observed that positive samples often alternated with negative samples throughout the trial and that the evolution of the number of vRNA copies per time interval was unpredictable, which is in contradiction to the observations made in experiment 1. Sera from three chickens were found positive at 14 and 21 dpe (Table 2). Consequently, 50% of susceptible SPF chickens were positive by rRT–PCR without showing seroconversion, which contradicts what was observed in experiment 1.
In the SE group, all chickens were positive by rRT–PCR as well. In this exposure group, 28/48 OP swabs, analysed between 0 and 14 dpe, were positive. A total of 16/48 CL swabs were found positive during that same period, which is much more than was observed in the DWE group. Quantities of vRNA were larger in OP swabs than in CL swabs. Compared to the DWE group, OP swabs contained similar quantities of vRNA, but CL swabs contained significantly larger quantities of vRNA (Table 1). Positive swabs often alternated with negative swabs as well. However, none of the SPF chicken serum samples had detectable antibodies directed against viral NP (Table 2).
Trial 3b
In trial 3b, only one of the three inoculated ducks was successfully infected; at the time of removing the ducks from the trial and establishing the DWE and SE groups, no vRNA was detected in the drinking water, which had a clean appearance, and the floor swab contained only 103,5 vRNA copies/ml storage medium. After exposure, all swabs taken from the susceptible SPF chickens were negative by rRT–PCR for both exposure groups and none of the susceptible SPF chickens seroconverted.
Assessment of environmental infection pressure
In the DWE group from trial 3, further follow-up of the drinking water showed that vRNA was still present for at least 14 more days and that the quantities of vRNA declined slowly. Testing these samples for virus viability revealed that all drinking-water samples contained viable virus and that the titres declined along with the amounts of vRNA (Table 3). In this trial's SE group, follow-up of vRNA in floor swabs showed that quantities of vRNA had decreased markedly by 1 dpe and then remained more or less constant until 14 dpe at a level which was ± 3 log10 below the initial concentration of vRNA. Testing virus viability showed that only some floor swabs contained viable virus and that the virus titres of these samples were generally low (Table 3). A comparison between vRNA quantities and TCID50 titres in drinking-water and floor samples collected during the first week post-exposure was made possible by calculating the AUC values for these samples (Table 3). These results show that 1 TCID50 in drinking-water samples corresponded to roughly 400 vRNA copies, whereas 1 TCID50 in floor swabs corresponded to roughly 50 000 vRNA copies.
DISCUSSION
In this study, we studied the mechanisms of the introduction of a duck-originated H5N3 LPAIV to poultry. The strain H5N3 A/Anas platyrhynchos/09–884/2008 was selected for this study because characterization had previously revealed that it is part of a highly dynamic population of wild-bird LPAIVs that are circulating in wild birds in Belgium. Moreover, genetic characterization of the virus, conducted by Van Borm et al. has indicated that no additional glycosylation sites are present in the HA and that the length of the NA stalk is not reduced, indicating that this strain is most likely not adapted to poultry [Reference Van14]. Results from our assessment of the infectivity of this virus in SPF chickens and Pekin ducks provide additional proof that this strain is phenotypically adapted to ducks and that a high infectious dose may be necessary to cause active infection in SPF chickens [Reference Swayne and Slemons22]. Moreover, we observed that the profile of infectivity was different in Pekin ducks than in SPF chickens. Indeed, we observed seroconversion together with virus shedding in 5/12 inoculated SPF chickens and seroconversion without virus shedding in 4/12 oculo-nasally inoculated SPF chickens. The remaining three inoculated SPF chickens exhibited neither immune response nor virus shedding. The most important virus shedding route for inoculated SPF chickens was through the oropharynx while CL virus shedding was negligible. Virus shedding by the CL route was much stronger in ducks than in SPF chickens and was always associated with seroconversion.
In a second step, in order to assess the introduction potential of H5N3 A/Anas platyrhynchos/09–884/2008 to poultry by its natural host, we conducted a series of transmission experiments with inoculated Pekin ducks and susceptible SPF chickens. From these results, it was shown that co-housing of susceptible SPF chickens and inoculated Pekin ducks led to seroconversion in a similar percentage of SPF chickens compared to oculo-nasal inoculation. However, in these experiments, all SPF chickens were positive by rRT–PCR for both OP and CL swabs, even those that did not seroconvert. This discordancy may possibly be explained by the occurrence of localized virus replication which is detectable in the oropharynges and cloacae and which the chickens were able to clear with a T-cell-mediated response, without developing a systemically measurable humoral immune response. However, to our knowledge, no scientific account exists on the occurrence of localized influenza virus replication without the development of antibodies in veterinary medicine. Alternatively, some rRT–PCR results for chicken swabs may have come from the ingestion of virus from the environment rather than from a replicative infection. Since we observed a strong contamination of the isolators with infective duck faeces, it is possible that scratching and pecking the floor or just lying down caused large doses of virus from the environment to end up in the chickens' beaks or cloacae. Unfortunately, the impact of this environmental contamination on our results is impossible to estimate. Combining the two possibilities, we assumed that the positive swabs obtained from SPF chickens reflected both virus shedding and virus exposure. The existence of a significant relationship between OP and CL chicken swabs and environmental samples might therefore indicate that a higher environmental contamination leads to a higher exposure and possibly higher virus shedding. Furthermore, the significant relationship between duck CL swabs and environmental samples indicates the importance of CL virus shedding by infectious ducks for the build-up of environmental infection pressure.
Since the occurrence of a LPAIV infection without the development of antibodies is not certain and since the use of serology reduces the possibility of incorrectly interpreting truly non-infected SPF chickens as infected, we decided to consider the immune response of chickens as a decisive factor for our model. The estimates we obtained for β dc indicate that close contact between a single duck infected with H5N3 A/Anas platyrhynchos/09–884/2008 and fully susceptible chickens may result in 0·3 new infection/day. Since a previous study suggests that transmission of this virus between chickens does not occur [Reference Claes19], we can conclude that this LPAIV can be transmitted from wild birds to poultry through close contact, but that subsequent circulation is unlikely. However, other H5/H7 LPAIVs of wild-bird origin have been shown to have a strong infectious potential for chickens [Reference Marche23]. Therefore, it can be expected that such wild bird-originated LPAIVs may have the ability to circulate within chicken populations after introduction. Conducting similar interspecies transmission experiments as the one presented in the present study, but with a wild bird-originated H5/H7 LPAIV which has better infectious potential for poultry or which is closely related to a poultry-adapted strain may provide clarity on this matter.
It should be borne in mind that the experimental design of our transmission experiment, involving close contact, created an artificial environment which may have substantially forced transmission and that the results must be somehow mitigated. Indeed, in existing situations where contact between poultry and wild birds may occur, wild ducks and chickens do not tend to come into such close contact with each other for such a long period of time. An observational study conducted by Welby et al. (unpublished data) has demonstrated ducks visiting smallholder poultry flocks may repose inside the holding, eat and make use of the drinking water for only a few minutes up to a few hours. This leads to faecal contamination of the soil, the surroundings of the feeding systems and the drinking water, whereas close contact might be somewhat limited because of more space. Since LPAIV-infected ducks tend to shed virus primarily through the intestinal tract [Reference Vandalen24] and since environmental contamination has been postulated to play an important role in several transmission experiments [Reference Claes19, Reference Vandalen24–Reference Joh27], we designed and conducted our animal experiment to force the system and examine the dynamics of LPAIV introduction through the most similar wild bird-induced environmental contamination.
Two types of environmental transmission were considered; transmission through contaminated drinking water and transmission through contaminated surfaces. Results from this experiment proved that drinking water contaminated by faeces from three LPAIV-shedding ducks can lead to detection of vRNA in all, and seroconversion in 50% of chickens exposed to it. These results prove that drinking water contaminated by wild birds is an efficient route for the introduction of LPAIVs into poultry holdings. Moreover, since ducks may shed up to 109 EID50/g faeces [28], the initial virus concentration of 106·2 TCID50/ml reported for drinking water in the present study can reasonably be reached with a small degree of faecal contamination.
When chickens were exposed to contaminated surfaces, vRNA was detected in the oropharynges and cloacae but seroconversion was not observed. Therefore, this route of transmission appears less effective than drinking water, probably because water is a more virus-friendly environment. Indeed, it is well-known that AIVs survive well in humid environments [Reference Stallknecht16, Reference Brown29–Reference Leung32], whereas survival in faeces or on surfaces may be compromised, especially at low humidity [Reference De Benedictis, Beato and Capua33–Reference Shortridge36]. Therefore, we additionally performed an endpoint titration on CEFs to estimate TCID50/ml sample medium for environmental samples. This test considers live virus instead of vRNA, which may also come from defective viruses, and thus allows the assessment of virus viability in drinking water or on surfaces for this study. Since these results indicate that the proportion of vRNA copies/TCID50 per ml is much larger for the SE group than for the DWE group, it can be concluded that a large proportion of vRNA in the former is in fact derived from defective viruses which did not participate in the build-up of infection pressure [Reference Guan37].
Alternatively, water may well be a better vehicle for infecting chickens with wild bird-originated LPAIVs than surface contamination is. Indeed, besides being ingested, water droplets can also be inhaled when drinking, which leads to virus being directly delivered to the respiratory epithelium, similar to what occurs in an intranasal or intratracheal inoculation. On the other hand, the primary way through which virus on faecally contaminated feed or surfaces can be taken in by chickens is through ingestion. The virus is thus only delivered to the oral, pharyngeal or oesophageal epithelium, which are less reactive than the respiratory epithelium [Reference Hirabayashi38]. Finally, the survival of the virus in the drinking water containing different (amounts of) disinfectants should be investigated further, as this could represent an additional preventive measure.
We have demonstrated that LPAIV-shedding ducks may build-up an infection pressure that is strong enough to cause intake of large amounts of virus in exposed chickens. While a highly sensitive method such as rRT–PCR was effective in detecting exposure and environmental contamination, this method did not appear useful for assessing whether virus intake led to a true infection in chickens or not. Therefore, in order to avoid overestimation of virus transmission in experiments involving a strong degree of environmental contamination, we recommend the use of other methods for determining infection in animals. The use of serology could be useful; however, this method may be less sensitive as localized virus replication may not always induce a systemic immune response. We additionally provided evidence that drinking water contaminated with faeces from LPAIV-shedding ducks alone, may cause successful infection of poultry with a wild-bird LPAIV. However, since the virus used in the present study is not easily transmitted between chickens [Reference Claes19], the chances of such an event leading to circulation of the virus in poultry appear limited. It is therefore possible that some of these introduction events remain unnoticed. However, other wild bird-originated LPAIVs may have good infective characteristics for poultry [Reference Swayne and Slemons22] and may thus be more prone to be transmitted in chickens. If such a virus is introduced efficiently, this may lead to the establishment of the virus in poultry and to the possible emergence of a HPAIV.
ACKNOWLEDGEMENTS
The authors thank Mieke Steensels, Anouar Nadi, Eva Ngabirano, Christophe Delgrange and Marc van den Broeck for their technical and laboratory assistance. This work was supported by Belgian Contractual Research: Flutree (grant RF6217) and Flutrans (Belspo).
DECLARATION OF INTEREST
None.






