


INTRODUCTION
Williams syndrome (WS) is a rare neurodevelopmental disorder caused by a microdeletion in the q11.23 region of chromosome 7. The incidence rate of its typical forms is 1/7000 to 1/25,000 live births (Bellugi, Lichtenberger, Mills, Galaburda, & Korenberg, Reference Bellugi, Lichtenberger, Mills, Galaburda and Korenberg1999; Mervis & Morris, Reference Mervis and Morris2007; Meyer-Linderberg, Mervis, & Berman, Reference Meyer-Linderberg, Mervis and Berman2006). The main clinical manifestations of WS are: (1) characteristic facial features (e.g., small upturned nose, long philtrum, wide mouth, and small chin); (2) supravalvular aortic stenosis (SVAS) and/or peripheral pulmonary artery stenosis (PPAS); and (3) an atypical neuropsychological phenotype characterized by a moderate to severe intellectual disability, severe impairment of visuospatial function, mild language impairment (Ferrero et al., Reference Ferrero, Howald, Micale, Biamino, Augello, Fusco and Merla2010), and hypersociability (Bellugi et al., Reference Bellugi, Lichtenberger, Mills, Galaburda and Korenberg1999). Most commonly (91–95%), WS is a result of deletion of 1.5 megabases (Mb) spanning a total of 24 genes. Near the middle of this deletion span is ELN, the gene that encodes elastin, mutations of which are associated with the SVAS/PPAS aspect of the syndrome (Mervis & Morris, Reference Mervis and Morris2007; Osborne, Reference Osborne2010; Porter et al., Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012).
The WS neuropsychological profile is strongly associated with a severe impairment in visuospatial capacity (Capirci, Sabbadini, & Volterra, Reference Capirci, Sabbadini and Volterra1996; Garayzábal, Reference Garayzábal2005; Garayzábal & Cuetos, Reference Garayzábal and Cuetos2008). This visuospatial impairment could be consequent to the loss of genes involved in the structure and functions of the parietal cortex and the so-called dorsal stream of processing thought to carry location and motion information anterolaterally from primary visual cortex to parietal areas (Atkinson et al., Reference Atkinson, Anker, Braddick, Nokes, Mason and Braddick2001; Atkinson & Braddick, Reference Atkinson and Braddick2012; Atkinson & Nardini, Reference Atkinson and Nardini2008; García-Nonell, Rigau-Ratera, Artigas-Pallarés, García-Sánchez, & Estévez-González, Reference García-Nonell, Rigau-Ratera, Artigas-Pallarés, García-Sánchez and Estévez-González2003). Some authors have found gray matter volume alterations or reductions in the dorsal occipitoparietal sulcus/vertical region of the intraparietal sulcus, which could explain, at least in part, dorsal stream vulnerability in WS (Atkinson, Reference Atkinson2017; Atkinson & Braddick, Reference Atkinson and Braddick2011; Meyer-Linderberg et al., Reference Meyer-Linderberg, Kohn, Mervis, Kippenhan, Olsen, Morris and Berman2004).
There has been increasing recognition of the variability in WS cognitive phenotype, including variability in intelligence (Tassabehji et al., Reference Tassabehji, Metcalfe, Karmiloff-Smith, Carette, Grant, Dennis and Donnai1999), executive functions (Morris et al., Reference Morris, Mervis, Hobart, Gregg, Bertrand, Ensing and Stock2003), the expression of autistic-like features (Edelmann et al., Reference Edelmann, Prosnitz, Pardo, Bhatt, Cohen, Lauriat and McInnes2007), and visuospatial abilities (Karmiloff-Smith et al., Reference Karmiloff-Smith, Grant, Ewing, Carette, Metcalfe, Donnai and Tassabehji2003). Advancements in genetic and molecular biology techniques, such as chromosomal microarrays (CMAs), can enable genomic alterations to be identified with higher precision than previously. Hence, the cognitive phenotype can be analyzed in combination with CMA data to determine the genes affected by each patient’s deletion and thus help to explain the variability observed in the neuropsychological phenotypes of WS patients.
Studies conducted in animal models, as well as in patients with atypical deletions, have identified genes within the range of the WS microdeletion that could affect the neuropsychological phenotype of WS, including, from centromere to telomere (see Figure 1), FZD9, BAZ1B, STX1A, LIMK1, CLIP2, GTF2IRD1, and GTF2I (Botta et al., Reference Botta, Novelli, Mari, Sabani, Korenberg, Osborne and Dallapiccola1999; Frangiskakis et al., Reference Frangiskakis, Ewart, Morris, Mervis, Bertrand, Robinson and Keating1996; Gray, Karmiloff-Smith, Funnell, & Tassabehji, Reference Gray, Karmiloff-Smith, Funnell and Tassabehji2006; Morris et al., Reference Morris, Mervis, Hobart, Gregg, Bertrand, Ensing and Stock2003; Osborne, Reference Osborne2010; Vandeweyer, Van der Aa, Reyniers, & Kooy, Reference Vandeweyer, Van der Aa, Reyniers and Kooy2012; Wang, Spörle, Paperna, Schughart, & Francke, Reference Wang, Spörle, Paperna, Schughart and Francke1999). These genes are known regulators of proteins related to localized structural and functional brain development (Broadbent et al., Reference Broadbent, Farran, Chin, Metcalfe, Tassabehji, Turnpenny and Karmiloff-Smith2014; Gao et al., Reference Gao, Bellugi, Dai, Mills, Sobel, Lange and Korenberg2010; Karmiloff-Smith et al., Reference Karmiloff-Smith, Broadbent, Farran, Longhi, D’Souza, Metcalfe and Sansbury2012, Reference Karmiloff-Smith, Grant, Ewing, Carette, Metcalfe, Donnai and Tassabehji2003; Osborne, Reference Osborne2010; Tassabehji et al., Reference Tassabehji, Metcalfe, Karmiloff-Smith, Carette, Grant, Dennis and Donnai1999; Vandeweyer et al., Reference Vandeweyer, Van der Aa, Reyniers and Kooy2012).
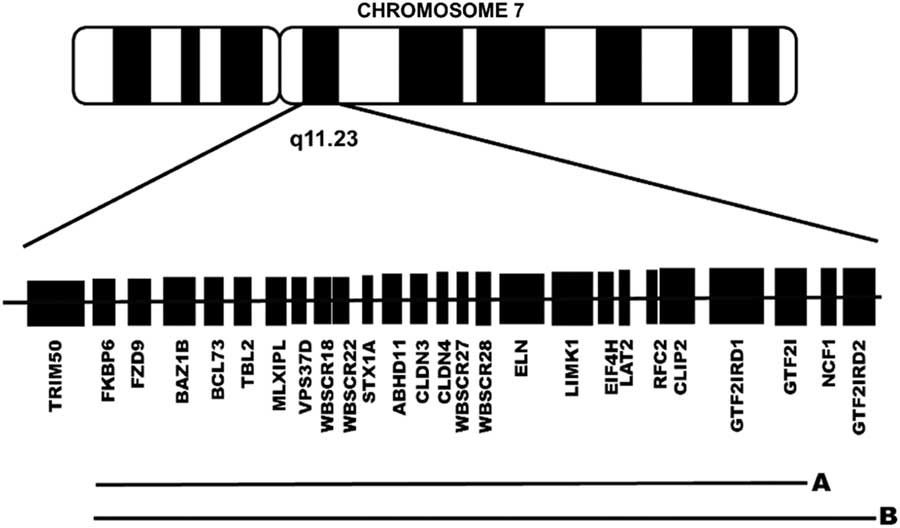
Fig 1 Schematic representation of the genes lost in the typical group of patients with a preserved GTF2IRD2 (1.5 Mb deletion) (A) and in the atypical group of patients without GTF2IRD2 (1.8 Mb deletion) (B).
Most cases of WS (91–95%) are associated with loss of GTF2I and GTF2IRD1, which are normally expressed in the cerebellum, hippocampus (Chailangkarn, Noree, & Muotri, Reference Chailangkarn, Noree and Muotri2018), and intraparietal sulcus (Hoeft et al., Reference Hoeft, Dai, Haas, Sheau, Mimura, Mills and Reiss2014). Disruption of these genes has been associated with reduced fear and aggression (Young et al., Reference Young, Lipina, Tam, Mandel, Clapcote, Bechard and Osborne2008), altered visuomotor integration and visuospatial processing (Hirota et al., Reference Hirota, Matsuoka, Chen, Salandanan, Lincoln, Rose and Korenberg2003; Hoeft et al., Reference Hoeft, Dai, Haas, Sheau, Mimura, Mills and Reiss2014), intellectual disability (Morris et al., Reference Morris, Mervis, Hobart, Gregg, Bertrand, Ensing and Stock2003), and highly sociable behavior (Chailangkarn et al., Reference Chailangkarn, Noree and Muotri2018; Young et al., Reference Young, Lipina, Tam, Mandel, Clapcote, Bechard and Osborne2008).
A small minority (5–8%) of patients with WS present with an atypical 1.8-Mb deletion that encompasses two additional genes, including GTF2IRD2, a member of the GTF2I gene family, whose members encode transcription factors required for synapse development and neurulation (Allen Brain Atlas, 2010; Makeyev et al., Reference Makeyev, Erdenechimeg, Mungunsukh, Roth, Enkhmandakh, Ruddle and Bayarsaihan2004; Tipney et al., Reference Tipney, Hinsley, Brass, Metcalfe, Donnai and Tassabehji2004; Uhlén et al., Reference Uhlén, Fagerberg, Hallström, Lindskog, Oksvold, Mardinoglu and Fredrik2015). The GTF2I gene family also includes BEN, a TFII-I family gene (Li et al., Reference Li, Huang, Luo, Huang, Lin and Fang2015). In mice, haploinsufficiency of TFII-I proteins results in a variety of phenotypic manifestations, including embryonic lethality and brain hemorrhage, as well as vasculogenic, craniofacial, and neural tube defects (Enkhmandakh et al., Reference Enkhmandakh, Makeyev, Erdenechimeg, Ruddle, Chimge, Tussie-Luna and Bayarsaihan2009).
GTF2IRD2 mRNA expression levels are high in the cerebral cortex, specifically in the prefrontal and parietal areas, as well as in the cerebellum (Allen Brain Atlas, 2010; Porter et al., Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012; Tipney et al., Reference Tipney, Hinsley, Brass, Metcalfe, Donnai and Tassabehji2004; Uhlén et al., Reference Uhlén, Fagerberg, Hallström, Lindskog, Oksvold, Mardinoglu and Fredrik2015). The protein encoded by GTF2IRD2 is also a TFII-I family protein (Makeyev et al., Reference Makeyev, Erdenechimeg, Mungunsukh, Roth, Enkhmandakh, Ruddle and Bayarsaihan2004) expressed in the brain (Tipney et al., Reference Tipney, Hinsley, Brass, Metcalfe, Donnai and Tassabehji2004), specifically in parietal, frontal (orbitofrontal and dorsolateral) cortices and in the cerebellum (Allen Brain Atlas, 2010; Porter et al., Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012; Uhlén et al., Reference Uhlén, Fagerberg, Hallström, Lindskog, Oksvold, Mardinoglu and Fredrik2015). GTF2IRD2 has been shown to regulate the activity of GTF2IRD1 and other TFII-I proteins during the postnatal period by direct interaction and sequestration of the proteins in a nuclear compartment (Palmer et al., Reference Palmer, Taylor, Santucci, Widago, Chan, Yeo and Hardeman2012).
The aforementioned evidence indicates that disruption of GTF2I family genes in WS is associated with altered social behavior, negative emotionality, visuospatial ability, and intellectual ability (Crespi & Hurd, Reference Crespi and Hurd2014; Hoeft et al., Reference Hoeft, Dai, Haas, Sheau, Mimura, Mills and Reiss2014; Morris et al., Reference Morris, Mervis, Hobart, Gregg, Bertrand, Ensing and Stock2003; Young et al., Reference Young, Lipina, Tam, Mandel, Clapcote, Bechard and Osborne2008). Meanwhile, GTF2IRD2, specifically, has been associated with visuospatial functioning, social reasoning, and cognitive flexibility (Porter et al., Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012). Therefore, we hypothesized that loss of GTF2IRD2 may underlie visuospatial and social skill impairment severity in WS patients. To test this hypothesis in this study, we evaluated the cognitive, behavioral, and adaptive effects of GTF2IRD2 deletion in WS patients.
METHODS
Participants
Twelve patients (7 males) diagnosed with WS (age range, 7–18 years) were recruited from the Asociación Nacional de Síndrome de Williams A.C., the Asociación Viviendo con Síndrome de Williams A.C., and from the Genetics Service of the “Dr. Eduardo Liceaga” General Hospital of Mexico. All patients were living in the metropolitan area of Mexico City. Before starting the assessments, the parents of the participants signed written informed consent forms, and the patients’ participation was approved by the ethics committee of the Faculty of Higher Education Iztacala, which adheres to the Helsinki Declaration.
Based on the CMA results (described below), patients were divided into a typical GTF2IRD2-retained group and an atypical GTF2IRD2-deleted group (Figure 1). The typical group consisted of 8 patients (5 males) with a 1.5-Mb microdeletion and a chronological age (CA) mean of 11.5 years (SD, 3.59), including five who were in a special education program and four who had a perinatal complication. The atypical group consisted of 4 patients (2 males) with a 1.8-Mb microdeletion and a mean CA of 12 years (SD, 4.97), including one who was in a special education program, one who had a perinatal complication. None of the patients in either the typical group or the atypical group were taking medication. As shown in Supplementary Table 1S, the two groups were similar with respect to CA, mental age, full-scale IQ (FSIQ), and number of completed years of education (Mann Whitney U, all p > .05). Exact Fisher tests revealed no significant between-group differences with respect to gender constitution (p = .57), special education rate (p = .27), and perinatal complication rate (p = .42).
Setting and Procedures
A geneticist conducted a clinical assessment of each participant and evaluated his or her CMA results. A trained neuropsychologist conducted a neuropsychological evaluation in two 1.5-hr sessions in a cubicle at the Genetics Services of the “Dr. Eduardo Liceaga” General Hospital of Mexico. The CMA results were revealed to the neuropsychologist after all neuropsychological tests were completed. All assessments were conducted in Spanish, and Spanish-language neuropsychological instruments were used.
Genetic Assessment
In the geneticist’s office, a blood sample was obtained from each participant for CMA Cyto-Scan Optima analysis to determine his or her 7q11.23 microdeletion scope. This genetic analysis technique is based on nucleic acid hybridization and fluorescence analysis of imaged chromosomes (Venegas Vega, Reference Venegas Vega2012).
Neuropsychological Instruments
To obtain each participant’s clinical history, a structured interview was carried out to collect information about each patient’s personal (pathological and non-pathological), hereditary-familial, and developmental history. The Wechsler Intelligence Scale for Children, 4th edition (WISC-IV; Wechsler, Reference Wechsler2007) and the Wechsler Adult Intelligence Scale, 4th edition (WAIS-IV; Wechsler, Reference Wechsler2014) were administered to evaluate the intelligence of patients who were ≤17 years of age and of patients who were 17–18 years of age, respectively. The following WISC-IV/WAIS-IV scales were considered: Verbal Comprehension Index (VCI), Perceptual Reasoning Index (PRI), Working Memory Index (WMI) and Processing Speed Index (PSI), and FSIQ. Attention, memory, and executive function were evaluated with the NEUROPSI Attention and Memory subtests (Ostrosky-Solís, Guevara-López, & Matute, Reference Ostrosky-Solís, Guevara-López and Matute2012) and the Child Neuropsychological Assessment 2 (Evaluación Neuropsicológica Infantil, ENI-2) (Matute, Rosselli, & Ardilla, Reference Matute, Rosselli and Ardila2014). Visual-spatial processing ability was assessed with the Developmental Test of Visual Perception, 3rd edition (DTVP3), which uses the Frostig perception evaluation method (Hammill, Pearson, & Voress, Reference Hammill, Pearson and Voress2016).
To detect a wide range of emotional and behavioral problems, parents of the WS patients completed the family questionnaire of the Assessment System for Children and Adolescents (Sistema de Evaluación de Niños y Adolescentes (SENA) (Fernández-Pinto, Santamaría, Sánchez-Sánchez, Carrasco, & Del Barrio, Reference Fernández-Pinto, Santamaría, Sánchez-Sánchez, Carrasco and Del Barrio2015). The SENA measures internalizing problems (e.g., depression, anxiety, social anxiety), externalizing problems (e.g., hyperactivity and impulsivity, attention problems, aggressiveness), specific problems (e.g., developmental delays, learning disabilities), areas of vulnerability, and psychological resources. Finally, parents also completed the Adaptive Behavior Assessment System, 2nd edition (ABAS-II) parent form (Harrison & Oakland, Reference Harrison and Oakland2008); the ABAS-II provides a complete assessment of adaptive skills across the lifespan.
Statistical Analysis
Mann Whitney U tests were used to evaluate intergroup differences in neuropsychological test scores because the cohort was too small to produce parametric datasets. Due to this study’s the small size, no p-level adjustment for multiple comparisons was made. Nevertheless, we calculated Cohen’s d statistical values for each between-group comparison result, such that d = 0.2 was considered to represent a small effect, 0.5 a medium effect, and 0.8 a large effect. We analyzed the standardized scores obtained on all administered neuropsychological instruments.
RESULTS
Participants in both groups obtained FSIQ scores in the range of 40 to 60. No intergroup differences were found in any of the WISC-IV/WAIS-IV indices, nor in the NEUROPSI attention and memory test or the ENI-2 Executive Function scale. The two groups had similar verbal ability scores. On the other hand, a tendency toward lower cognitive performance was observed in the atypical GTF2IRD2-deleted group, relative to the typical group, especially in the tests that assess cognitive flexibility. The results of the intergroup comparison can be seen in Supplementary Tables S2–S4.
In DTVP-3 (Table 1), the typical group (
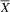 ${\overline X} $
= 3.5; SD = 0.84) performed better on the Visual Closure subtest than the atypical group (
${\overline X} $
= 3.5; SD = 0.84) performed better on the Visual Closure subtest than the atypical group (
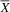 ${\overline X} $
= 2.00; SD = 0.82), with a large effect size (U = 2; p = .04; d = 1.83). The typical group (
${\overline X} $
= 2.00; SD = 0.82), with a large effect size (U = 2; p = .04; d = 1.83). The typical group (
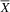 ${\overline X} $
= 10.83; SD = 1.17) showed a non-significant trend toward performing better than the atypical group (
${\overline X} $
= 10.83; SD = 1.17) showed a non-significant trend toward performing better than the atypical group (
 ${\overline X} $
= 7.5; SD = 3.32) on the Form Consistency subtest, with a large effect size (U = 3; p = .07; d = 1.53). Relative to the typical group (
${\overline X} $
= 7.5; SD = 3.32) on the Form Consistency subtest, with a large effect size (U = 3; p = .07; d = 1.53). Relative to the typical group (
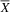 ${\overline X} $
= 85; SD = 4.34), the atypical group (
${\overline X} $
= 85; SD = 4.34), the atypical group (
 ${\overline X} $
= 73.75; SD = 9.95) had greater alterations in the Reduced Motor Response Index, of which the Visual Closure and Form Consistency subtests are components, with a large effect size (U = 2.5; p = .04; d = 1.67). Note that for this index perceptual abilities are highly weighted given the limited use of motor skills in the execution of its subtests.
${\overline X} $
= 73.75; SD = 9.95) had greater alterations in the Reduced Motor Response Index, of which the Visual Closure and Form Consistency subtests are components, with a large effect size (U = 2.5; p = .04; d = 1.67). Note that for this index perceptual abilities are highly weighted given the limited use of motor skills in the execution of its subtests.
Table 1 Comparison of DTVP-3 between atypical and typical WS group

Note: Two participants did not perform the DTVP-3 because they did not return.
a Large effect size.
In the SENA inventory, the atypical group (
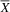 ${\overline X} $
= 80.75; SD = 10.72) scored higher than the typical group (
${\overline X} $
= 80.75; SD = 10.72) scored higher than the typical group (
 ${\overline X} $
= 59.75; SD = 15.58) on the Isolation scale, with a large effect size (U = 4; p = .04; d = 1.46), suggesting that atypical patients spend more time alone (Table 2). In the same way, the typical group (
${\overline X} $
= 59.75; SD = 15.58) on the Isolation scale, with a large effect size (U = 4; p = .04; d = 1.46), suggesting that atypical patients spend more time alone (Table 2). In the same way, the typical group (
 ${\overline X} $
= 52.88; SD = 9.51) performed better than the atypical group (
${\overline X} $
= 52.88; SD = 9.51) performed better than the atypical group (
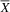 ${\overline X} $
= 36.25; SD = 3.40) on the Emotional Intelligence scale of the SENA, with a large effect size (U = 1; p = .01; d = 2.17), evidencing better social skills and behaviors of the typical group compared to the atypical group. Analyzing the items that make up the SENA Emotional Intelligence and Isolation scales, we found that, compared to the typical group, the atypical group present greater deficiencies in several constructs of social cognition (Adolphs, Reference Adolphs2009), including emotional recognition (e.g., “When I feel bad, he/she notices”), empathy (e.g., “Pay attention to how others feel”), and theory of mind (e.g., “Knows how to put themselves in the another’s place”).
${\overline X} $
= 36.25; SD = 3.40) on the Emotional Intelligence scale of the SENA, with a large effect size (U = 1; p = .01; d = 2.17), evidencing better social skills and behaviors of the typical group compared to the atypical group. Analyzing the items that make up the SENA Emotional Intelligence and Isolation scales, we found that, compared to the typical group, the atypical group present greater deficiencies in several constructs of social cognition (Adolphs, Reference Adolphs2009), including emotional recognition (e.g., “When I feel bad, he/she notices”), empathy (e.g., “Pay attention to how others feel”), and theory of mind (e.g., “Knows how to put themselves in the another’s place”).
Table 2 Comparison of SENA between atypical and typical WS group

a Large effect size.
Finally, the typical group (
 ${\overline X} $
= 6.75; SD = 3.06) also performed significantly better than the atypical group (
${\overline X} $
= 6.75; SD = 3.06) also performed significantly better than the atypical group (
 ${\overline X} $
= 2.25; SD = 2.50) on the ABAS-II Leisure scale, with a large effect size (U = 4; p = .04; d = 1.46). Furthermore, the typical group (
${\overline X} $
= 2.25; SD = 2.50) on the ABAS-II Leisure scale, with a large effect size (U = 4; p = .04; d = 1.46). Furthermore, the typical group (
 ${\overline X} $
= 8.88; SD = 2.10) also performed better on the ABAS-II Social scale than the atypical group (
${\overline X} $
= 8.88; SD = 2.10) also performed better on the ABAS-II Social scale than the atypical group (
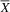 ${\overline X} $
= 5; SD = 2.71), with a large effect size (U = 3; p = .02; d = 1.65). The poorer social skills of the atypical group relative to the typical group are consistent with a greater deficit in social cognition, which could be related to the loss of GTF2IRD2 (Table 3).
${\overline X} $
= 5; SD = 2.71), with a large effect size (U = 3; p = .02; d = 1.65). The poorer social skills of the atypical group relative to the typical group are consistent with a greater deficit in social cognition, which could be related to the loss of GTF2IRD2 (Table 3).
Table 3 Comparison of ABAS-II between atypical and typical WS group

a Large effect size.
DISCUSSION
Our typical group with loss of GTF2I/GTF2IRD1 showed a neuropsychological phenotype characteristic of WS, whereas our atypical group, whose 1.8-Mb deletion resulted in the additional loss of GTF2IRD2, exhibited certain visuospatial/perception and social cognition alterations together with a reduced ability to socialize. GTF2IRD2 has been observed to have a genetic regulatory influence on GTF2I and GTF2IRD1 (Palmer et al., Reference Palmer, Taylor, Santucci, Widago, Chan, Yeo and Hardeman2012); all three of these genes are expressed in frontal and parietal cortices, and the cerebellum (Allen Brain Atlas, 2010; Porter et al., Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012; Tipney et al., Reference Tipney, Hinsley, Brass, Metcalfe, Donnai and Tassabehji2004; Uhlén et al., Reference Uhlén, Fagerberg, Hallström, Lindskog, Oksvold, Mardinoglu and Fredrik2015). Considering the present results, we posit that the loss of this group of genes, and the consequent loss of the availability of the proteins encoded by these genes in the prefrontal cortex, parietal cortex, and cerebellum, where they are normally highly expressed, led to greater cognitive impairments than is typical of WS.
We found that both groups had similar cognitive-behavioral phenotypes with respect to verbal abilities. This sparing of verbal abilities may be related to the low-density of expression of GTF2IRD2 in temporal areas (Allen Brain Atlas, 2010; Colantuoni et al., Reference Colantuoni, Lipska, Hyde, Tao, Leek, Colantuoni and Kleinman2011; Uhlén et al., Reference Uhlén, Fagerberg, Hallström, Lindskog, Oksvold, Mardinoglu and Fredrik2015; Zhang et al., Reference Zhang, Chen, Sloan, Bennett, Schoize, O’Keefe and Wu2014). Meanwhile, we found that GTF2IRD2 deletion in WS was associated with impairments in visuospatial skills and social cognition. These results suggest that GTF2IRD2 may contribute to the severity of the neuropsychological phenotype of WS, especially with respect to deficiencies in some visuospatial abilities and some aspects of social cognition.
Concerning visuospatial ability, patients with a GTF2IRD2 deletion showed reduced visual closure capacity, compared to patients in whom the gene was conserved. This ability is related to recognition of how components of a unit are associated to form a figure (Frostig, Reference Frostig1999). Formerly, Porter et al. (Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012) found low scores on spatial tasks in nine WS patients who lost this gene. However, in the present study, we found specifically that visual closure ability, form consistency, and figure-grounding, tasks that do not include a motor component and that form the reduced motor response index, are weaknesses in these patients. These findings support the notion that the GTF2IRD2 deletion phenotype may have more of a perceptual component than a visual-motor one.
Additionally, visual closure capacity has been found to be related to global attention tasks (D’Souza, Booth, Connolly, Happe, & Karmiloff-Smith, Reference D’Souza, Booth, Connolly, Happe and Karmiloff-Smith2016; Porter & Coltheart, Reference Porter and Coltheart2006). Dorsal stream vulnerability theory encompasses visual closure capacity together with navigation capacity, use of spatial labels, and visuoconstructive (Atkinson, Reference Atkinson2017; Atkinson et al., Reference Atkinson, Anker, Braddick, Nokes, Mason and Braddick2001; Atkinson & Braddick, Reference Atkinson and Braddick2011, Reference Atkinson and Braddick2012; Atkinson & Nardini, Reference Atkinson and Nardini2008). According to this theory, there are two perceptual networks: (1) the ventral stream responsible for object and face recognition, which is retained in patients with WS; and (2) the dorsal stream associated with object location and spatial management (Atkinson, Reference Atkinson2017).
Several studies have provided evidence suggesting that the dorsal stream is related to occipito-parietal networks, specifically the intraparietal sulcus, precuneus, and inferior and superior parietal gyrus (Kravitz, Saleem, Baker, & Mishkin, Reference Kravitz, Saleem, Baker and Mishkin2011; Meyer-Linderberg et al., Reference Meyer-Linderberg, Kohn, Mervis, Kippenhan, Olsen, Morris and Berman2004). Global ability and closure have also been reported to be related to intraparietal sulcus alterations (Atkinson, Reference Atkinson2017; Meyer-Linderberg et al., Reference Meyer-Linderberg, Kohn, Mervis, Kippenhan, Olsen, Morris and Berman2004; Mobbs et al., Reference Mobbs, Eckert, Menon, Mills, Korenberg, Galaburda and Reiss2007). Our finding of impaired visual closure in WS patients without GTF2IRD2 are consistent with prior findings showing that, like patients with right parietal lobe lesions, WS patients can identify and copy the elements of visual stimuli, but fail to integrate them into a whole (global process) (Atkinson et al., Reference Atkinson, Anker, Braddick, Nokes, Mason and Braddick2001; Atkinson & Braddick, Reference Atkinson and Braddick2012; Atkinson & Nardini, Reference Atkinson and Nardini2008). Hence, dorsal stream vulnerability theory (Atkinson & Nardini, Reference Atkinson and Nardini2008) could explain the greater visuospatial alterations in people with WS associated with loss of GTF2IRD2, relative to typical WS patients.
In this context, it is noteworthy that both the parietal cortex and cerebellum, areas with unusually high GTF2IRD2 expression (Allen Brain Atlas, 2010; Porter et al., Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012; Uhlén et al., Reference Uhlén, Fagerberg, Hallström, Lindskog, Oksvold, Mardinoglu and Fredrik2015), are involved in visuospatial ability and are altered markedly in WS (Martens, Wilson, & Reutens, Reference Martens, Wilson and Reutens2008). The parietal and prefrontal cortices in the right hemisphere have been associated with the recognition of emotions in faces, judgment about them, theory of mind, and empathy (Adolphs, Reference Adolphs2009; Shamay-Tsoory, Tomer, Goldsher, Berger, & Aharon-Peretz, Reference Shamay-Tsoory, Tomer, Goldsher, Berger and Aharon-Peretz2004). Structural and functional alterations have been found in the amygdala and right orbital-medial prefrontal cortex of WS patients performing tasks that evaluate these domains of social cognition (Bellugi et al., Reference Bellugi, Järvinen-Pasley, Doyle, Reilly, Reiss and Korenberg2007; Mimura et al., Reference Mimura, Hoeft, Kato, Kobayashi, Sheau, Piggot and Reiss2010). WS patients perform similarly to patients with right parietal damage on visuospatial tasks (Atkinson et al., Reference Atkinson, Anker, Braddick, Nokes, Mason and Braddick2001; Atkinson & Braddick, Reference Atkinson and Braddick2012; Atkinson & Nardini, Reference Atkinson and Nardini2008); this greater deficit may be associated with lower visuospatial and social performance in those patients with a GTF2IRD2 deletion.
Regarding behavioral profile, our finding that patients with a GTF2IRD2 deletion presented several behavioral problems associated with social cognition indicate considerable challenges for these patients in appropriate expression of empathy, recognition of the emotional state of another, and theory of mind. This finding is consistent with previous results of Porter et al. (Reference Porter, Dobson-Stone, Kwok, Schofield, Beckett and Tassabehji2012), who identified more difficulties in theory of mind tasks in patients with lost, versus retained, GTF2IRD2. Likewise, individual case reports of WS patients with co-deletion of GTF2I, GTF2IRD1, and GTF2IRD2 described predominant autistic features (Edelmann et al., Reference Edelmann, Prosnitz, Pardo, Bhatt, Cohen, Lauriat and McInnes2007; Karmiloff-Smith et al. Reference Karmiloff-Smith, Broadbent, Farran, Longhi, D’Souza, Metcalfe and Sansbury2012). Conversely, our adaptive skills scale results may indicate better social abilities performance and leisure activities in WS patients with conserved GTF2IRD2, and their relatively superior performance in these areas may be related to the preservation of their ability to perceive others’ emotions and their more typical performance on tests of theory of mind and empathy.
Limitations
It is important to emphasize that in this study we did not apply specific or experimental tests that assessed the patients’ cognitive-emotional abilities directly. However, the results of the inventories completed by the parents had moderate to large effect sizes. The limitations of this study include: (1) a lack of neuroimaging studies, which could identify structural inter-hemispheric differences (especially in the parietal and frontal cortices and in the cerebellum) associated with visuospatial processes and social cognition; (2) a lack of psychological and experimental tests that assess the components of social cognition directly; and (3) a small sample, which precludes a strong statistical analysis and limits the generalizability of the results. Notwithstanding, this work provides an approach to examining the relationship among genes, brain, cognition, and behavior in WS patients.
CONCLUSION
The present findings provide new evidence of the importance of GTF2IRD2 in the cognitive, behavioral, and adaptive phenotypes of WS patients. The results support the notion that the locally elevated expression of GTF2IRD2 in the right prefrontal and parietal cortices, when the gene is present, reflect its role in the cognitive, behavioral, and adaptive functions that are impaired in WS patients with deletion of GTF2IRD2. Furthermore, given GTF2IRD2’s regulatory influence on GTF2IRD1, loss of GTF2IRD2 may result in structural alterations in sites where GTF2IRD1 is normally expressed, leading to more profound effects on visuospatial and social skills. It is necessary to continue studying the phenotypic outcomes associated with different deletion variants in WS patients to identify the neuropsychological influence of specific genes affected in WS.
ACKNOWLEDGMENTS
All authors involved in this investigation declare no conflicts of interest. PAPIIT IA301916 and CONACYT CVU 478060 supported this work. We thank The Asociación Nacional de Síndrome de Williams A.C., The Asociación Viviendo con Síndrome de Williams A.C., and Genetics Service of the “Dr. Eduardo Liceaga” General Hospital of Mexico. Dr. Hermelinda Salgado-Ceballos thanks the IMSS Foundation for the scholarship of excellence received.
Supplementary materials
To view supplementary material for this article, please visit https://doi.org/10.1017/S1355617718000711






