


Some selective serotonin reuptake inhibitors (SSRIs) and benzodiazepines, and the serotonin-noradrenaline reuptake inhibitor venlafaxine, are efficacious in generalised anxiety disorder (Reference Baldwin and PolkinghornBaldwin & Polkinghorn, 2005). Placebo-controlled double-blind studies demonstrate the efficacy of the SSRIs paroxetine (Reference Pollack, Zaninelli and GoddardPollack et al, 2001; Reference Rickels, Zaninelli and McCaffertyRickels et al, 2003), sertraline (Reference Allgulander, Dahl and AustinAllgulander et al, 2004; Reference Brawman-Mintzer, Knapp and RynnBrawman-Mintzer et al, 2006), and escitalopram (Reference Davidson, Bose and KorotzerDavidson et al, 2004; Reference Goodman, Bose and WangGoodman et al, 2005). A 24-week study found escitalopram (10-20 mg/day) and paroxetine (20-50 mg/day) to have similar efficacy (Reference Bielski, Bose and ChangBielski et al, 2005). These studies provide no clear evidence for a dose-response relationship. Current guidelines for assessing efficacy recommend a minimum length of 8 weeks, using a placebo-controlled design comprising fixed doses to establish optimal dosage (Reference Montgomery and van Zwieten-BootMontgomery & van Zwieten-Boot, 2002). This study aimed to compare the efficacy of fixed doses of escitalopram (5, 10 or 20 mg/day) with placebo over 12 weeks' treatment, including paroxetine (20 mg/day) as an active reference.
METHOD
Study design and closing schedule
This randomised, placebo-controlled, fixeddose, active-reference study included 63 centres in 10 countries. It was conducted in accordance with the principles of Good Clinical Practice (ICH, 1996) and the Declaration of Helsinki (World Medical Association, 2000). Local ethics committees approved the study design, and eligible patients gave their written informed consent before participating. After screening, patients entered a 1-week, single-blind, placebo lead-in period before being randomised to 12 weeks of double-blind treatment with fixed doses of escitalopram (5, 10 or 20 mg/day), paroxetine (20 mg/day), or placebo. Patients who completed double-blind treatment entered a randomised staggered 2-week (1-week doubleblind, then 1-week single-blind) placebo wash-out period. Efficacy and tolerability were assessed at baseline and after 1, 2, 4, 6, 8, 10, 12, 13 and 14 weeks; a safety follow-up visit was performed 14 days after the last wash-out visit.
Allocation to treatment
Study medications were capsules for oral administration, of identical appearance, taste and smell. The oxalate salt of escitalopram was used in the capsules. Patients who met selection criteria at the screening and baseline visits were assigned to 12 weeks of double-blind treatment in a 1:1:1:1:1 ratio of 5 mg escitalopram to 10 mg escitalopram to 20 mg escitalopram to 20 mg paroxetine to placebo according to a computer-generated randomisation list drawn up by H. Lundbeck A/S. The timing of down-titration for the 5 mg and 10 mg escitalopram groups and the 20 mg paroxetine group was built into the overall randomisation scheme; patients in these groups were assigned to continue current active treatment or start placebo wash-out treatment at week 13 in a 1:1 ratio of active treatment to placebo. The details of the randomisation series were unknown to any of the investigators and were contained in a set of sealed opaque envelopes. At each study centre, sequentially enrolled patients were assigned the lowest randomisation number available in blocks of ten. All study personnel and participants were masked to treatment assignment for the duration of the study.
Patient population
The selection criteria were chosen to select physically healthy male and female out-patients with a primary diagnosis of generalised anxiety disorder according to DSM-IV-TR (American Psychiatric Association, 2000) criteria. The Mini International Neuropsychiatric Interview (MINI: Reference Sheehan, Lecrubier and SheehanSheehan et al, 1998) was used to establish the diagnosis and to confirm the presence or absence of other disorders. Patients aged 18-65 years old with a Hamilton Anxiety Scale (HAMA; Reference HamiltonHamilton, 1959) total score ⩾20, and a score of ⩾2 on both HAMA item 1 (anxious mood) and item 2 (tension) at screening and at baseline could be included. A low level of depressive symptoms was allowed using the Montgomery-Åsberg Depression Rating Scale (MADRS; Reference Montgomery and ÅsbergMontgomery & Åsberg, 1979), i.e. total score ⩽16 at screening and at baseline.
Patients with the following disorders within the previous 6 months (based on DSM-IV-TR criteria, confirmed using the MINI) were excluded: major depressive disorder, panic disorder, social anxiety disorder, post-traumatic stress disorder, bipolar disorder, obsessive-compulsive disorder, eating disorders, body dysmorphic disorder, substance misuse disorder, any personality disorder that could jeopardise the evaluation of the treatment for primary generalised anxiety, as judged by the investigator, and any current or previous psychotic disorder as defined by DSM-IV-TR. Patients were also excluded if they were at risk of suicide (according to the investigator's judgement), had a score >3 on item 10 (suicidal thoughts) of the MADRS, or had made a serious suicide attempt within the past year or were receiving cognitive-behavioural therapy, electroconvulsive therapy, cognitive therapy or problem-solving treatment, or planned to initiate such therapy. Furthermore, patients with an unstable serious illness and/or serious sequelae of liver or renal insufficiency, or cardiac, vascular, pulmonary, gastrointestinal, endocrine, neurological, infectious, neoplastic or metabolic disturbance were also excluded. Patients were excluded if they had taken psychoactive substances, anxiolytics, antidepressants, monoamine oxidase inhibitors, benzodiazepines, β-blockers (use of anti-hypertensives other than β-blockers was permitted as long as the dose had been stable for 6 months and remained fixed during the study), tryptophan, oral antipsychotics, narcotic analgesics (except intermittent use of codeine-based analgesics), warfarin sodium, digitalis, cardiac glycosides, type 1c anti-arrhythmics, phenytoin, cimetidine, regular daily therapy with any hypnotic (except zolpidem, zopiclone, or zaleplon for insomnia, but not more than 3 times per week), psychoactive herbal remedies, antiepileptics, ongoing prophylactic treatment with lithium, valproate or carbamazepine, and triptans within the 2 weeks before the screening visit, and any investigational drug or depot antipsychotics within 6 months before the screening visit.
Efficacy assessments
The primary end-point was defined as the adjusted mean change in HAMA total score from baseline to week 12, based on the intention-to-treat set and using last-observation-carried-forward last-observation-carried-forward analysis.
Secondary efficacy measures included: change from baseline in HAMA total score at each visit; Clinical Global Impression - Severity (CGI-S) and Clinical Global Impression - Improvement (CGI-I; Reference GuyGuy, 1976) score per visit; proportion of responders per visit using two criteria (⩾50% reduction in HAMA total score compared with baseline, and CGI-I score of 1 and 2); proportion of remitters (HAMA total score ⩽7) per visit; and change from baseline in the self-rating Hospital Anxiety and Depression Scale (HAD; Reference Zigmond and SnaithZigmond & Snaith, 1983) anxiety sub-scale score at weeks 6 and 12. The investigators were trained by a physician experienced in the use of HAMA before inclusion of patients into the study to increase interrater reliability. Patient ratings were conducted by the same person at each visit, whenever possible.
Tolerability assessments
Tolerability was based on the incidence of adverse events throughout the study. The Discontinuation Emergent Signs and Symptoms (DESS) scale is a 43-item checklist (Reference Rosenbaum, Fava and HoogRosenbaum et al, 1998) designed to assess possible treatment-related discontinuation symptoms. For this study, the DESS was slightly modified to include four extra items, reported after stopping SSRI treatment: vivid dreams, electric shock-like sensations, somnolence, and feeling tense. An event was considered discontinuation emergent if it appeared during the previous 7 days, or if a previously reported event had worsened. The modified DESS was assessed at week 12 and during the wash-out period (weeks 13 and 14) for patients who had completed the 12-week double-blind treatment period. Unresolved symptoms in the DESS checklist were subject to enquiry at the safety follow-up visit. Half of the patients randomised to escitalopram 5 or 10 mg/day, or 20 mg/day paroxetine, received placebo during the 2-week washout period, whereas the other half continued on active treatment for 1 week (week 13) and received placebo for the second week (week 14). Patients who were randomised to 20 mg escitalopram were down-titrated to 10 mg escitalopram for 1 week (week 13) before they received placebo (week 14).
Statistical analysis
A minimum of 130 patients in each treatment group (intention-to-treat) was expected to provide a standardised effect size of 0.35, that is a significant treatment difference from placebo of at least 35% of the pooled standard deviation when comparing the mean change from baseline to week 12 (last observation carried forward) in HAMA total score, using a two-sided t-test with 80% power at a 5% level of significance.
All efficacy analyses were conducted on the intention-to-treat population consisting of all randomised patients who took at least one dose of double-blind study medication and who had at least one valid post-baseline assessment of the HAMA. The prospectively defined primary efficacy end-point was the adjusted mean change from baseline in HAMA total score at week 12, based on intention to treat (last observation carried forward). Comparisons of the primary efficacy end-point between escitalopram and placebo were made using analysis of covariance (ANCOVA) with treatment and centre as fixed factors, and with the baseline HAMA total score as a covariate. To adjust for multiple testing, an F-test was used to test the overall null hypothesis of equal mean changes in the three escitalopram groups and the placebo group. If the overall F-test was significant at the 5% level, pairwise comparisons of each of the three escitalopram dose groups and the placebo group were made using two-sided t-tests with the overall mean square error as the error term at a 5% level of significance. Likewise, paroxetine was compared pairwise with the other treatment groups using two-sided t-tests with the overall mean square error as the error term at a 5% level of significance.
The secondary efficacy analyses of mean change from baseline to each visit in the HAMA total scores and HAD sub-scale score were analysed by ANCOVA (observed cases and last observation carried forward) using the model described for the primary analysis. The CGI-I scores were analysed using analysis of variance. Between-group comparisons of patients considered to be treatment responders and between patients considered to be remitters were carried out using Fisher's exact test.
Incidences of adverse events were compared between treatment groups using Fisher's exact test based on all randomised patients who took at least one dose of double-blind medication.
The modified DESS total scores during the wash-out period were analysed for patients completing the study based on observed cases using ANCOVA with treatment and centre as factors, and the modified DESS total score at the start of the wash-out period as a covariate.
RESULTS
Patient baseline characteristics
There were no clinically relevant differences between groups in patient demographic or clinical characteristics at baseline (Table 1). The small differences between groups in HAMA total score at baseline are unlikely to be of clinical significance, and are adjusted for in the primary efficacy analysis by the inclusion of baseline score as a covariate. Most patients were Caucasian, and there was an approximately 2:1 ratio of women to men, with a mean age of about 41 years. Baseline HAMA, CGI-S and MADRS scores indicated a moderately to severely ill patient population with a low level of depressive symptoms.
Table 1 Baseline patient characteristics

| PBO | ESC 5 mg | ESC 10 mg | ESC 20 mg | PAR 20 mg | |
|---|---|---|---|---|---|
| Patients randomised, n 1 | 139 | 134 | 136 | 133 | 140 |
| Patients treated, n | 139 | 134 | 136 | 133 | 139 |
| Women, n (%) | 93 (67) | 78 (58) | 91 (67) | 92 (69) | 84 (60) |
| Age, years | |||||
| Mean (s.d.) | 41.8 (11.6) | 40.7 (11.9) | 41.8 (12.8) | 41.0 (12.2) | 41.7 (12.0) |
| Range | 19–64 | 18–65 | 19–65 | 19–65 | 18–64 |
| Caucasian, n (%) | 138 (99.3) | 132 (98.5) | 135 (99.3) | 131 (98.5) | 137 (98.6) |
| Efficacy scores 2 | |||||
| HAMA total score (s.d.) | 27.1 (4.6) | 27.1 (4.5) | 26.0 (4.1) | 27.7 (4.9) | 27.3 (4.2) |
| HAD anxiety sub-scale score (s.d.) | 13.5 (3.6) | 13.1 (3.4) | 13.1 (3.7) | 13.6 (3.3) | 13.0 (3.0) |
| CGI–S (s.d.) | 4.6 (0.7) | 4.6 (0.8) | 4.5 (0.7) | 4.6 (0.7) | 4.6 (0.7) |
| MADRS total score (s.d.) | 11.4 (3.2) | 11.2 (3.0) | 11.0 (3.1) | 11.4 (3.0) | 11.0 (3.1) |
CGI–S, Clinical Global Impression – Severity; ESC, escitalopram; HAD, Hospital Anxiety and Depression Scale; HAMA, Hamilton Rating Scale for Anxiety; MADRS, Montgomery–Åsberg Depression Rating Scale; PAR, paroxetine; PBO, placebo
1. Randomised patients per country: Czech Republic, 62; Denmark, 75; Estonia, 84; Finland, 107; France, 44; Germany, 67; Holland, 4; Norway, 54; Spain, 44; UK, 141; total, 682
2. Based on intention-to-treat population (see Fig. 1)

Fig. 1 Patient disposition for the 14-week study period. ESC, escitalopram (5, 10 or 20 mg); ITT, intention-to-treat; PAR, paroxetine; PBO, placebo.
Withdrawals from the study
Fig. 1 shows the patient disposition for the 14-week study period for all groups. A total of 98 patients (14%) withdrew from the study during the 12-week, double-blind period (Table 2), and withdrawal rates ranged from 10.8% to 18.7%.
Table 2 Withdrawals from study according to primary reason during the 12-week study period

| PBO | ESC 5 mg | ESC 10 mg | ESC 20 mg | PAR 20 mg | |
|---|---|---|---|---|---|
| Patients randomised, n | 139 | 134 | 136 | 133 | 140 |
| Patients treated, n | 139 | 134 | 136 | 133 | 139 |
| Patients withdrawn, n (%) | 15 (10.8) | 17 (12.7) | 18 (13.2) | 22 (16.5) | 26 (18.7) |
| Primary reason, n (%) | |||||
| Adverse event(s) | 4 (2.9) | 7 (5.2) | 8 (5.9) | 14 (10.5) * | 13 (9.4) * |
| Lack of efficacy | 5 (3.6) | 5 (3.7) | 0 (0) * | 2 (1.5) | 4 (2.9) |
| Withdrawal of consent | 4 (2.9) | 0 (0) | 1 (0.7) | 1 (0.8) | 3 (2.2) |
| Non-compliance | 1 (0.7) | 1 (0.7) | 2 (1.5) | 1 (0.8) | 0 (0) |
| Protocol violation | 1 (0.7) | 1 (0.7) | 5 (3.7) | 3 (2.3) | 4 (2.9) |
| Administrative or other | 0 (0) | 2 (1.5) | 0 (0) | 1 (0.8) | 1 (0.7) |
| Lost to follow-up | 0 (0) | 1 (0.7) | 2 (1.5) | 0 (0) | 1 (0.7) |
ESC, escitalopram; PAR, paroxetine; PBO, placebo
* P <0.05 v. placebo (χ2-test)
The proportion of patients that withdrew because of adverse events was relatively low (<11% in any treatment group and <7% overall). Compared with the placebo group, significantly more patients (chi-square test, P<0.05) in the escitalopram 20 mg and paroxetine 20 mg groups withdrew because of adverse events. Withdrawal rates due to lack of efficacy in the escitalopram 5 and 20 mg, paroxetine 20 mg and placebo groups were comparable. Compared with placebo, significantly fewer patients in the escitalopram 10 mg group withdrew because of lack of efficacy.
Primary efficacy analysis
The prospectively defined primary efficacy end-point (adjusted mean change in HAMA total score from baseline to week 12, last observation carried forward) showed that treatment with escitalopram 10 and 20 mg was significantly superior to placebo at week 12 (Table 3 and Fig. 2). Escitalopram 5 mg and paroxetine 20 mg were not significantly superior to placebo at week 12.

Fig. 2 Mean change from baseline in Hamilton Rating Scale for Anxiety (HAMA) total scores by visit (intention-to-treat, observed cases) and at week 12, LOCF (last observation carried forward). Difference v. placebo, * P<0.05; ** P<0.01; *** P<0.001. Difference v. paroxetine # P<0.05 (analysis of covariance).
Table 3 Mean change from baseline to week 12 in HAMA total score (ITT, LOCF)

| Treatment | Difference v. placebo 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean change | s.e. | Difference | 95% CI | P | |||||
| PBO | 138 | –14.20 | 0.66 | |||||||
| ESC 5 mg | 134 | –15.49 | 0.67 | –1.29 | –3.13 to 0.54 | 0.165 | ||||
| ESC 10 mg | 134 | –16.76 | 0.68 | –2.56 | –4.40 to –0.73 | 0.006 | ||||
| ESC 20 mg | 132 | –16.35 | 0.68 | –2.15 | –3.99 to –0.31 | 0.022 | ||||
| PAR 20 mg | 136 | –14.71 | 0.67 | –0.51 | –2.33 to 1.32 | 0.585 | ||||
ESC, escitalopram; ITT, intention-to-treat; LOCF, last observation carried forward; PAR, paroxetine; PBO, placebo
1. Analysis of covariance
The mean HAMA total scores decreased in all treatment groups from approximately 27 at baseline to less than 13 at week 12. The last-observation-carried-forward analysis demonstrated that escitalopram 10 mg was also significantly (ANCOVA, P<0.05) superior to paroxetine 20 mg at week 12 (HAMA difference of -2.06; 95% CI -3.90 to -0.21). In observed cases analyses, all three doses of escitalopram were significantly more efficacious than placebo at week 12 (ANCOVA, P<0.05), and showed a clear dose-response relationship (difference from placebo; escitalopram 5 mg: -1.67 (95% CI -3.25 to -0.09; P<0.05), escitalopram 10 mg: -2.17 (95% CI -3.75 to -0.59; P<0.01), escitalopram 20 mg: -3.10 (95% CI -4.72 to -1.49; P<0.001), Fig. 2). Separation of active treatment from placebo was apparent from week 4 onwards for escitalopram 10 mg and 20 mg (ANCOVA, P<0.05). A statistically significant separation of paroxetine 20 mg from placebo was seen at week 10 (ANCOVA, P<0.05). Analysis revealed that escitalopram 20 mg was significantly (ANCOVA, P<0.05) superior to paroxetine 20 mg at week 12 (observed cases) (HAMA difference of -1.90; 95% CI -3.54 to -0.25).
Secondary efficacy analysis
CGI-Improvement scores
The mean CGI-I scores at each visit are shown in Fig. 3. In the observed cases analyses, separation of active treatment from placebo was statistically significant from week 2 onwards for escitalopram 10 and 20 mg (ANOVA, P<0.05), including week 12 (last observation carried forward). Escitalopram 5 mg was statistically significantly superior to placebo at weeks 10 and 12 (ANOVA, P<0.05, observed cases) but not at week 12 (last observation carried forward). Paroxetine 20 mg was statistically significantly superior to placebo at weeks 4, 8 and 10 (ANOVA, P<0.05, observed cases). Escitalopram 10 mg was significantly superior to paroxetine 20 mg at week 12 (ANOVA, P<0.05, last observation carried forward).

Fig. 3 Mean Clinical Global Impression - Improvement (CGI-I) score according to visit (intention to treat, observed cases) and at week 12, LOCF (last observation carried forward). Difference v. placebo, * P<0.05; ** P<0.01; *** P<0.001. Difference v. paroxetine, # P<0.05 (analysis of covariance).
Response
Response based on the ⩾50% reduction in HAMA total score criterion was analysed for each treatment group at each visit by observed cases and at week 12 by last observation carried forward (data not shown). Significant (Fisher's exact test, observed cases; P<0.05) superiority in response v. placebo (43%) was seen at week 6 for escitalopram 10 mg, and at weeks 10 and 12 (Fisher's exact test, P<0.05) for escitalopram 20 mg. Escitalopram 20 mg was significantly (80%; Fisher's exact test, P<0.05) superior to paroxetine 20 mg (68%) at week 12. In the last-observation-carried-forward analysis, escitalopram 10 mg was significantly (72%; Fisher's exact test, P<0.05) superior to paroxetine 20 mg at week 12 (60%). The observed cases response rate at week 12 for escitalopram 5 mg (75%) was not significantly different from placebo (67%).
For response based on CGI-I, escitalopram 10 mg was significantly (Fisher's exact test, P<0.05) superior to paroxetine 20 mg at weeks 2 (last observation carried forward; 41% v. 29% respectively, data not shown) and 12 (78% v. 66% respectively; Fig. 4). Significant superiority over placebo was seen from week 2 (observed cases) onwards for escitalopram 10 mg (Fisher's exact test, P<0.05; Fig. 4). The escitalopram 20 mg group showed significance (Fisher's exact test, P<0.05) over placebo from week 4 onwards. Response rates (observed cases) at week 12 were 69% (placebo), 77% (escitalopram 5 mg), 83% (escitalopram 10 mg), 84% (escitalopram 20 mg), and 76% (paroxetine 20 mg). The proportion of responders in the paroxetine 20 mg group was not significantly different from that in the placebo group at any visit.
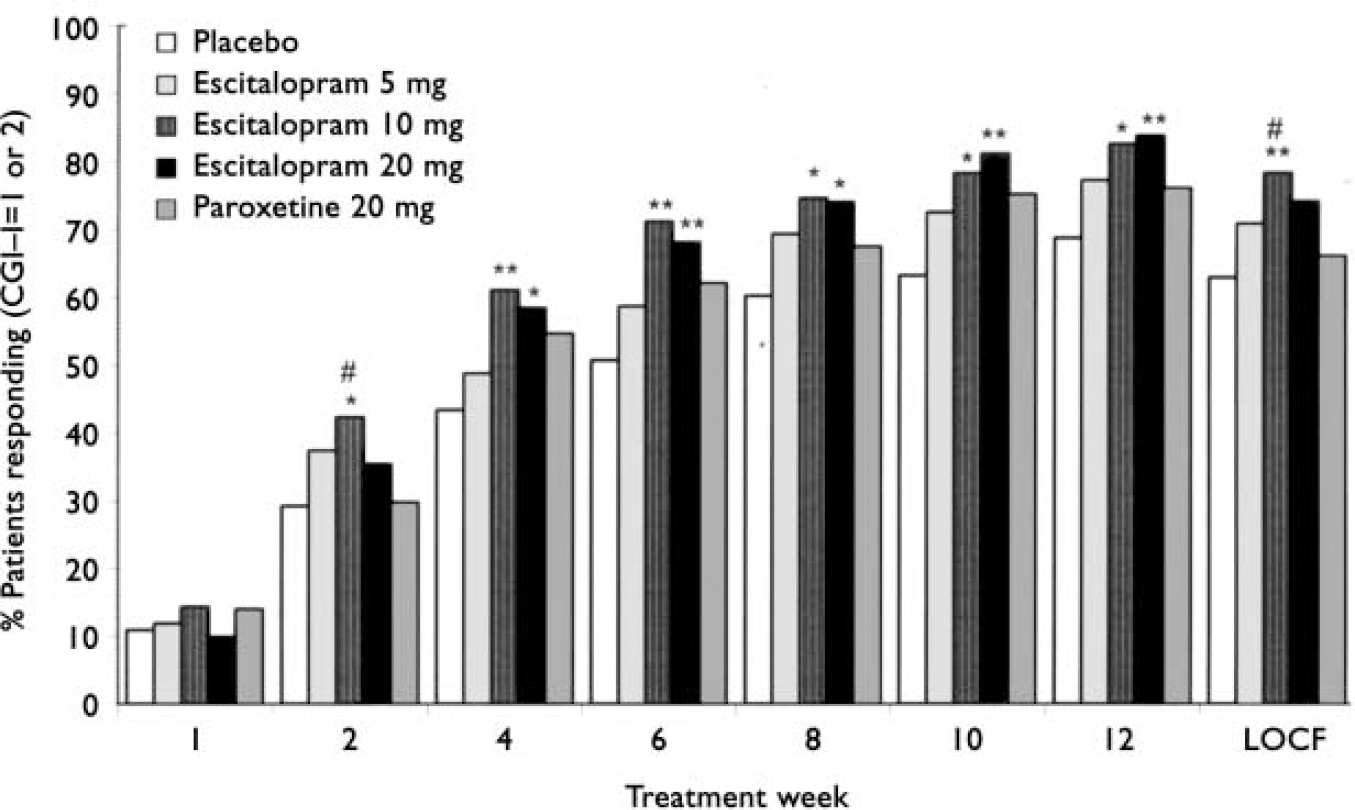
Fig. 4 Proportion of patients responding (defined as Clinical Global Impression - Improvement (CGI-I) score of 1 or 2) according to visit (intention to treat, observed cases) and at week 12, LOCF (last observation carried forward). Difference v. placebo, * P<0.05; ** P<0.01. Difference v. paroxetine, # P<0.05 (Fisher's exact test).
Remission
Remission was prospectively defined as a HAMA total score ⩽7 and was analysed for each treatment group at each visit by observed cases and at week 12 also by last observation carried forward. Superiority of escitalopram over placebo at week 12 was seen for all three doses of escitalopram (Fisher's exact test, P<0.05), and escitalopram 10 mg was significantly (48%; Fisher's exact test, P<0.05) superior to paroxetine 20 mg (33%). Superiority over placebo was seen at week 12 for escitalopram 5 mg and from week 8 onwards for escitalopram 10 and 20 mg (Fisher's exact test, observed cases, P<0.05; Fig. 5).

Fig. 5 Proportion of patients in remission (defined as a HAMA total score ⩽7) according to visit (intention-to-treat, observed cases) and at week 12, LOCF (last observation carried forward). Difference v. placebo, * P<0.05; ** P<0.01. Difference v. paroxetine, # P<0.05 (Fisher's exact test).
HAD anxiety sub-scale scores
In the analyses of the HAD anxiety subscale score, separation from placebo was statistically significant at both assessments (weeks 6 and 12) for escitalopram 10 and 20 mg (ANCOVA, P<0.05), whereas escitalopram 5 mg and paroxetine 20 mg were significantly superior to placebo only at week 6 (ANCOVA, P<0.05; Fig. 6). In the last-observation-carried-forward analysis, escitalopram 10 mg was furthermore significantly (ANCOVA, P<0.05) superior to paroxetine 20 mg at week 12.

Fig. 6 Mean change from baseline in Hospital Anxiety and Depression (HAD) anxiety sub-scale score (intention-to-treat, observed cases) and at week 12, LOCF (last observation carried forward). Difference v. placebo, * P<0.05; ** P<0.01; *** P<0.001. Difference v. paroxetine, # P<0.05 (analysis of covariance).
Tolerability
Adverse events
Table 4 shows adverse events with an incidence ⩾5% in any treatment group during the 12-week double-blind treatment period; there was no statistically significant difference in the number of patients experiencing adverse events across groups (chisquare test). The investigators considered the majority of the adverse events in all treatment groups to be mild or moderate. The approximate percentage of patients with adverse events considered to be related to study medication in each group was: 36% for placebo; 44% for escitalopram 5 mg; 54% for escitalopram 10 mg; 53% for escitalopram 20 mg; and 55% for paroxetine 20 mg. The incidence of fatigue, insomnia, diarrhoea, somnolence, increased sweating, yawning and anorgasmia were statistically significantly higher in at least one treatment group v. placebo (Fisher's exact test).
Table 4 Adverse events with an incidence of ≥5% according to group in the 12-week double-blind treatment period
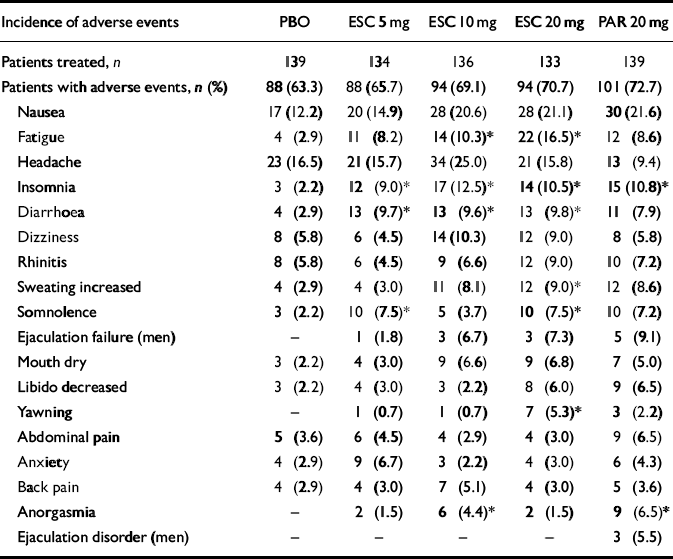
| Incidence of adverse events | PBO | ESC 5 mg | ESC 10 mg | ESC 20 mg | PAR 20 mg |
|---|---|---|---|---|---|
| Patients treated, n | 139 | 134 | 136 | 133 | 139 |
| Patients with adverse events, n (%) | 88 (63.3) | 88 (65.7) | 94 (69.1) | 94 (70.7) | 101 (72.7) |
| Nausea | 17 (12.2) | 20 (14.9) | 28 (20.6) | 28 (21.1) | 30 (21.6) |
| Fatigue | 4 (2.9) | 11 (8.2) | 14 (10.3)* | 22 (16.5)* | 12 (8.6) |
| Headache | 23 (16.5) | 21 (15.7) | 34 (25.0) | 21 (15.8) | 13 (9.4) |
| Insomnia | 3 (2.2) | 12 (9.0)* | 17 (12.5)* | 14 (10.5)* | 15 (10.8)* |
| Diarrhoea | 4 (2.9) | 13 (9.7)* | 13 (9.6)* | 13 (9.8)* | 11 (7.9) |
| Dizziness | 8 (5.8) | 6 (4.5) | 14 (10.3) | 12 (9.0) | 8 (5.8) |
| Rhinitis | 8 (5.8) | 6 (4.5) | 9 (6.6) | 12 (9.0) | 10 (7.2) |
| Sweating increased | 4 (2.9) | 4 (3.0) | 11 (8.1) | 12 (9.0)* | 12 (8.6) |
| Somnolence | 3 (2.2) | 10 (7.5)* | 5 (3.7) | 10 (7.5)* | 10 (7.2) |
| Ejaculation failure (men) | — | 1 (1.8) | 3 (6.7) | 3 (7.3) | 5 (9.1) |
| Mouth dry | 3 (2.2) | 4 (3.0) | 9 (6.6) | 9 (6.8) | 7 (5.0) |
| Libido decreased | 3 (2.2) | 4 (3.0) | 3 (2.2) | 8 (6.0) | 9 (6.5) |
| Yawning | — | 1 (0.7) | 1 (0.7) | 7 (5.3)* | 3 (2.2) |
| Abdominal pain | 5 (3.6) | 6 (4.5) | 4 (2.9) | 4 (3.0) | 9 (6.5) |
| Anxiety | 4 (2.9) | 9 (6.7) | 3 (2.2) | 4 (3.0) | 6 (4.3) |
| Back pain | 4 (2.9) | 4 (3.0) | 7 (5.1) | 4 (3.0) | 5 (3.6) |
| Anorgasmia | — | 2 (1.5) | 6 (4.4)* | 2 (1.5) | 9 (6.5)* |
| Ejaculation disorder (men) | — | — | — | — | 3 (5.5) |
ESC, escitalopram; PAR, paroxetine; PBO, placebo.
P <0.05 v. placebo (Fisher's exact test)
Table 5 shows adverse events with an incidence ⩾5% in any treatment group during the wash-out period. In each of the three escitalopram groups, the proportion of patients with adverse events during this period was not statistically different from that in the placebo group. The proportion of patients in the paroxetine 20 mg group with adverse events was significantly higher (chi-square test, P<0.01) than each of the other active treatment groups. The adverse events that had a statistically (Fisher's exact test, P<0.05) higher incidence in any active treatment group v. placebo during the wash-out period were: escitalopram 10 mg, insomnia (5.1%); escitalopram 20 mg, vertigo (3.6%); and paroxetine 20 mg, dizziness (19.5%), nausea (8.0%) and vertigo (5.3%). Dizziness had a statistically significantly higher incidence in the paroxetine 20 mg group than in any of the escitalopram groups.
Table 5 Adverse events with an incidence ≥5% according to group completing 12 weeks of treatment during the wash-out period

| Incidence of adverse events | PBO | ESC 5 mg | ESC 10 mg | ESC 20 mg | PAR 20 mg |
|---|---|---|---|---|---|
| Completers, n | 124 | 117 | 118 | 111 | 113 |
| Patients with adverse events, n (%) | 24 (19.4) | 13 (11.1) | 30 (25.4) | 21 (18.9) | 47 (41.6) * |
| Dizziness | 2 (1.6) | 1 (0.9) | 4 (3.4) | 4 (3.6) | 22 (19.5) * # |
| Headache | 8 (6.5) | 3 (2.6) | 6 (5.1) | 4 (3.6) | 6 (5.3) |
| Vertigo | 1 (0.8) | 4 (3.6) * | 6 (5.3) * | ||
| Insomnia | 6 (5.1) * | 2 (1.8) | 1 (0.9) | ||
| Nausea | 4 (3.4) | 1 (0.9) | 9 (8.0) * |
ESC, escitalopram; PAR, paroxetine; PBO, placebo
* P <0.05 v. placebo
# P <0.05 v. escitalopram (any dose) (Fisher's exact test)
Discontinuation Emergent Signs and Symptoms (DESS)
Fig. 7 shows the adjusted mean change from the start of the wash-out period in the total score on the modified DESS, as assessed by the DESS checklist. The dose received by participants in the escitalopram 20 mg group was down-tapered to 10 mg during week 13 and to placebo during week 14, without a randomised withdrawal design, and results are therefore not presented in Fig. 7. The mean total scores on the modified DESS were at a maximum after 7 days of wash-out treatment for the paroxetine 20 mg group and the 5 and 10 mg escitalopram groups. The mean change in the number of new or worsened DESS items was statistically significantly higher in the paroxetine 20 mg group than in the placebo group (4.2 v. 0.4; ANCOVA, P<0.001) at day 7. The discontinuation symptoms were transient and, after a further 7 days of wash-out treatment, returned to a level only slightly higher than that before starting wash-out treatment.

Fig. 7 Adjusted mean change from the start of the wash-out period in the modified Discontinuation Emergent Signs and Symptoms (DESS) total score (patients completed, observed cases). Difference v. placebo, *** P<0.001; difference v. paroxetine ### P<0.001 (analysis of covariance).
DISCUSSION
The aim of the current study was to examine three doses of escitalopram v. placebo and an active comparator (paroxetine) with proven efficacy for the medium-term (12 weeks) treatment of generalised anxiety disorder. The study design also allowed detailed evaluation of discontinuation symptoms, based on the DESS score. The baseline HAMA total score of approximately 27 and the baseline CGI-S score of approximately 4.5 indicate that this study population represents patients with moderate to severe illness.
There are a number of limitations to this study. First, the presence of the comorbid disorders typically found in patients with generalised anxiety disorder was low, as required by the protocol, and the results of this study are potentially less generalisable to samples seen in other clinical settings, although escitalopram has proven efficacy in major depression (Reference Burke, Gergel and BoseBurke et al, 2002; Reference Lepola, Loft and ReinesLepola et al, 2003), the most common comorbid disorder in generalised anxiety. Second, the placebo response rate (>60%) is high, when compared with previous flexible-dose or fixed-dose SSRI acute treatment studies in anxiety disorder, which report placebo response rates of 37% (flexible; Reference Allgulander, Dahl and AustinAllgulander et al, 2004), 38% (flexible; Reference Davidson, Bose and KorotzerDavidson et al, 2004), 46% (fixed; Reference Rickels, Zaninelli and McCaffertyRickels et al, 2003), 47% (flexible; Reference Pollack, Zaninelli and GoddardPollack et al, 2001) and 54% (flexible; Reference Brawman-Mintzer, Knapp and RynnBrawman-Mintzer et al, 2006). The high response rate to placebo in this investigation may result from the combination of the five-arm study design, the inclusion of multiple study centres and the frequency of study assessments. Third, there was no taper from 20 mg to 10 mg of paroxetine during the wash-out period, reflecting treatment recommendations at the time of the study. A fourth potential limitation lies within the method of analysis. It has been argued that last-observation-carried-forward analysis is not the best approach for evaluating data from randomised controlled trials (Reference White, Moodie and ThompsonWhite et al, 2003; Reference Everitt and WesselyEveritt & Wessely, 2004). All methods for the imputation of missing data have their limitations, but in disorders that do not deteriorate progressively, the conservative approach adopted in the above analysis is favoured by regulatory bodies, and was the specified form of data analysis in the study protocol.
The primary efficacy analysis (mean change from baseline in HAMA total score at week 12 using last observation carried forward) showed that escitalopram 10 and 20 mg were significantly superior to placebo. A dose-response relationship was seen at week 12 in the observed cases analysis. All three escitalopram doses were significantly superior to placebo; however, there was an increasing robustness of significance v. placebo from escitalopram 5 mg to 20 mg, which was associated with the highest response. Escitalopram 20 mg was also superior to paroxetine 20 mg in the observed cases analysis. The last-observation-carried-forward analysis demonstrated that escitalopram 10 mg was also significantly superior to paroxetine 20 mg at week 12. Mean HAMA and CGI-S scores decreased from week 10 to week 12, indicating that continued treatment might have resulted in further improvement, as found in a relapse-prevention study (Reference Allgulander, Florea and HuusomAllgulander et al, 2005), although only responders to acute treatment were eligible to continue in that study.
Paroxetine, 20 mg failed to show a significant difference from placebo in the primary efficacy analysis, which is probably attributable to the high response to placebo (Fig. 4). However, the decrease in HAMA from baseline to week 12 with paroxetine was approximately 14 points, numerically greater than placebo, and similar to that described in a previous study (Reference Rickels, Zaninelli and McCaffertyRickels et al, 2003), where paroxetine 20 mg for 8 weeks resulted in a 12.5-point reduction in the total HAMA score, and paroxetine 40 mg in a reduction of 12.2 points. In our study, paroxetine appeared efficacious on some of the secondary outcome measures: for example, it was significantly superior to placebo on the CGI-I at several points.
There were a number of secondary outcome measures, including the CGI-I, response and remission rates and the HAD anxiety sub-scale score. Using the CGI-I, in the observed cases analysis, the escitalopram 10 and 20 mg doses were superior to placebo from week 2 onwards, as well as at week 12 using last observation carried forward. According to response criteria, escitalopram 10 mg was superior to placebo from week 2 onwards and, at the end of the study, escitalopram 10 and 20 mg doses were significantly more efficacious than placebo. In the last-observation-carried-forward analysis, the response rate for the escitalopram 10 mg was also superior to paroxetine 20 mg. According to remission criteria, escitalopram 10 and 20 mg doses were more efficacious than placebo from week 8 onwards. Paroxetine 20 mg did not separate from placebo at any time point in this analysis. At study end, all three doses of escitalopram were superior to placebo and, in the last observation analysis, escitalopram 10 mg was superior to paroxetine 20 mg.
For the self-rating HAD anxiety subscale score, escitalopram 10 and 20 mg were significantly better than placebo at weeks 6 and 12. In the last-observation-carried-forward analysis, at 12 weeks escitalopram 10 mg was also superior to paroxetine 20 mg. This analysis showed a close agreement between the investigator's and the patient's assessment of treatment outcome.
Escitalopram 5 mg was not significantly superior to placebo across a variety of primary and secondary measures. This indicates that escitalopram 5 mg is probably too low a dose in this population of patients with generalised anxiety disorder. Higher doses are more efficacious, with an increased benefit for 20 mg, especially in terms of reaching symptomatic remission.
The incidence of adverse events during the 12-week study period was similar across all treatment groups. The proportion of patients with adverse events and withdrawals due to adverse events tended to increase as the dose of escitalopram increased. Only in the highest escitalopram dose group was there a significant difference in withdrawals due to adverse events compared with withdrawals with the placebo group, as was the case in the paroxetine 20 mg group. The adverse events that were reported during the 12-week treatment period for both escitalopram and paroxetine were characteristic for SSRIs. The incidence of patients reporting such events during the wash-out period was significantly higher in the paroxetine 20 mg group compared with escitalopram and placebo. The most frequent adverse event during wash-out was dizziness, which was reported by almost one-fifth of patients (22 out of 113) in the paroxetine group.
After 7 days of wash-out treatment, patients in the paroxetine 20 mg group were significantly more likely to have discontinuation-emergent symptoms compared with those in the placebo group. Patients who stopped treatment with escitalopram 10 mg had fewer discontinuation effects than those who stopped treatment with paroxetine 20 mg, this being consistent with the findings of a similar placebo-controlled study in social phobia (Reference Lader, Stender and BürgerLader et al, 2004). The discontinuation symptoms in the DESS checklist were transient and (after a further 7 days of wash-out treatment with placebo) returned to a level similar to that before patients started wash-out treatment. There was no significant difference between the escitalopram 5 mg and 10 mg groups v. placebo based on the modified DESS total score. In view of this, and the lower number of patients withdrawn because of adverse events in the escitalopram 10 mg group, clinicians may prefer to start with a 10 mg dose, increasing to 20 mg if patients show no signs of response after 4 weeks of treatment, when a significant difference from response to placebo was first seen in this study (Fig. 2).
In summary, escitalopram (10 and 20 mg/day) was efficacious and well tolerated in the medium-term treatment of generalised anxiety disorder. Escitalopram 10 mg was significantly more efficacious than paroxetine 20 mg.













eLetters
No eLetters have been published for this article.