



Introduction
Transition metal compounds that crystallize in the cubic B20-type structure belong to the space group P213 (# 198). One of the intriguing characteristics of a P213 space group is that it lacks the spatial parity operation of inversion (i.e., noncentrosymmetric). Many interesting properties emerge due to the inversion asymmetry, such as magnetoelectricity, second harmonic generation, pyroelectricity, ferroelectricity, and optical activity [Reference Shiv Halasyamani and Poeppelmeier1]. The main focus of this article is on chiral magnets in the bulk B20 crystal structure, which have been shown to host a peculiar spin texture in the form of a topologically stable skyrmion lattice. In this materials class, the skyrmion phase appears in a small pocket of the temperature–field phase diagram [Reference Banerjee, Rowland, Erten and Randeria2]. The origin of chiral spin modulation in noncentrosymmetric crystals is attributed to the energetic competition between the stronger magnetic exchange (J) interactions and the relatively weaker Dzyaloshinskii–Moriya (DM) interactions. An additional contributing factor to DM interaction is induced by the relativistic spin–orbit coupling that scales roughly with Z 2, where Z is the atomic number of the chemical element [Reference Shanavas, Popović and Satpathy3].
Skyrmions are regarded as promising for spintronics applications for at least three reasons [Reference Jan4]: (i) nanometer size, (ii) topological protection, and (iii) appearance of spin transfer torque, where magnetic structures and textures are manipulated by electric currents. Typical length of a magnetic domain wall that is used for hard disk recording is of the order of 30 nm [Reference Chappert, Fert and Van Dau5]; however, sub-30 nm size skyrmions are experimentally observed [Reference Kanazawa, Seki and Tokura6, Reference Caretta, Mann, Büttner, Ueda, Pfau, Günther, Hessing, Churikova, Klose, Schneider, Engel, Marcus, Bono, Bagschik, Eisebitt and Beach7], thus showing the exciting potential for encoding vast amount of information relative to the magnetic domain wall technologies [Reference Jan4]. The topological protection contributes to superior thermal stability [Reference Fert, Cros and Sampaio8, Reference Cortés-Ortuño, Wang, Beg, Pepper, Bisotti, Carey, Vousden, Kluyver, Hovorka and Fangohr9]. The spin transfer torque appears when the current density exceeds ∼106 A/m2, which is 4–5 orders of magnitude smaller than the current density required to move the ferroelectric domain walls [Reference Jonietz, Muhlbauer, Pfleiderer, Neubauer, Munzer, Bauer, Adams, Georgii, Boni, Duine, Everschor, Garst and Rosch10]. However, not all chiral magnets that form stable skyrmions satisfy the stringent property requirements for use in spintronics applications.
In Fig. 1, a plot is shown that summarizes the experimentally measured helical period (denoting skyrmion size) and helical transition temperature (T C) of several bulk B20 alloys [Reference Nagaosa and Tokura11]. It is interesting to note that none of the known B20 alloys satisfy the combined requirement of smaller helical period (<50 nm) and higher T C (>300 K) for practical spintronics applications. The best-known material in the B20 family is FeGe, which has a helical period and T C of 70 nm and 278 K, respectively. To date, only ∼20 compositions have been experimentally explored in the B20 alloy family. This indicates the challenge and the low-throughput nature of the synthesis and characterization efforts in the materials discovery cycle. The low-throughput nature of data generation from experiments is justified by the need for performing expensive and time-consuming synthesis (sometimes requiring nonequilibrium processing routes [Reference Zhang, Han, Ge, Du, Jin, Wei, Fan, Zhang, Li and Zhang12, Reference Stolt, Sigelko, Mathur and Jin13]). Moreover, nontrivial characterization studies are also needed to reliably report the crystal structure and magnetic properties of these alloys, adding to the challenge. However, the periodic table and the principles of alloy theory offer more avenues for composition design than what has been explored so far in the literature. We ask the following question—Are there novel B20 alloy compositions in the unexplored search space with T C better than that of known materials? Alloy design using computational methods have not attracted a lot of attention. Most of the computational efforts that involve density functional theory (DFT) calculations have focused on either providing an explanation of the observed properties from the viewpoint of the band theory or extracting properties such as the strength of the DM term as a function of alloy substitutions and epitaxial strain [Reference Gayles, Freimuth, Schena, Lani, Mavropoulos, Duine, Blügel, Sinova and Mokrousov14, Reference Mankovsky, Wimmer, Polesya and Ebert15, Reference Koretsune, Kikuchi and Arita16, Reference Pshenay-Severin and Burkov17, Reference Choi, Lin and Zhu18, Reference Shanavas and Satpathy19, Reference Karhu, Kahwaji, Monchesky, Parsons, Robertson and Maunders20, Reference Koretsune, Nagaosa and Arita21, Reference Kanazawa, Shibata and Tokura22]. In this work, a novel computational approach, built on the foundations of machine learning (ML) and DFT, is developed to accelerate the design of B20-based chiral magnets with improved T C. Although ML methods have been used in the past to predict the ferromagnetic Curie temperature of alloys [Reference Sanvito, Corey, Xue, Tiwari, Zic, Archer, Tozman, Venkatesan, Coey and Curtarolo23, Reference Nelson and Sanvito24], properties of hard permanent magnets [Reference Möller, Körner, Krugel, Urban and Elsässer25], two-dimensional materials [Reference Rhone, Chen, Desai, Yacoby and Kaxiras26] and magnetic properties of single-molecule magnets [Reference Holleis, Shivaram and Balachandran27, Reference Chi Dam, Pham, Ho, Nguyen and Nguyen28], no a priori rules exist that link alloy compositions to T C for the B20 alloys.

Figure 1: A summary of experimentally determined helical period in nanometer (x-axis) and helical transition temperature in K (T C, y-axis) of bulk B20 alloys taken from the published literature [Reference Nagaosa and Tokura11]. None of the known B20 alloys satisfy the combined requirement of smaller helical period (<50 nm) and higher T C (>300 K) for practical spintronics applications, which is denoted as “Target” in the figure. In this study, the focus is on the computational design of new B20 alloys that have T C > 300 K.
The objectives are accomplished by developing a data-driven approach that synergistically integrates insights from DFT calculations and published experimental literature with regression-based ML methods. The role of DFT is 2-fold. (i) To perform initial screening and identify promising alloying elements for ML. (ii) To validate the predictions from ML and provide a physical basis for meaningful interpretation of the ML outcome. The task of ML methods is to establish a quantitative relationship between the alloy compositions and the experimentally measured T C of known B20 alloys. The trained ML models are then used to rapidly predict promising new alloy compositions. The final outcome is the recommendation of new B20 chiral magnets for experimental validation.
Results and discussion
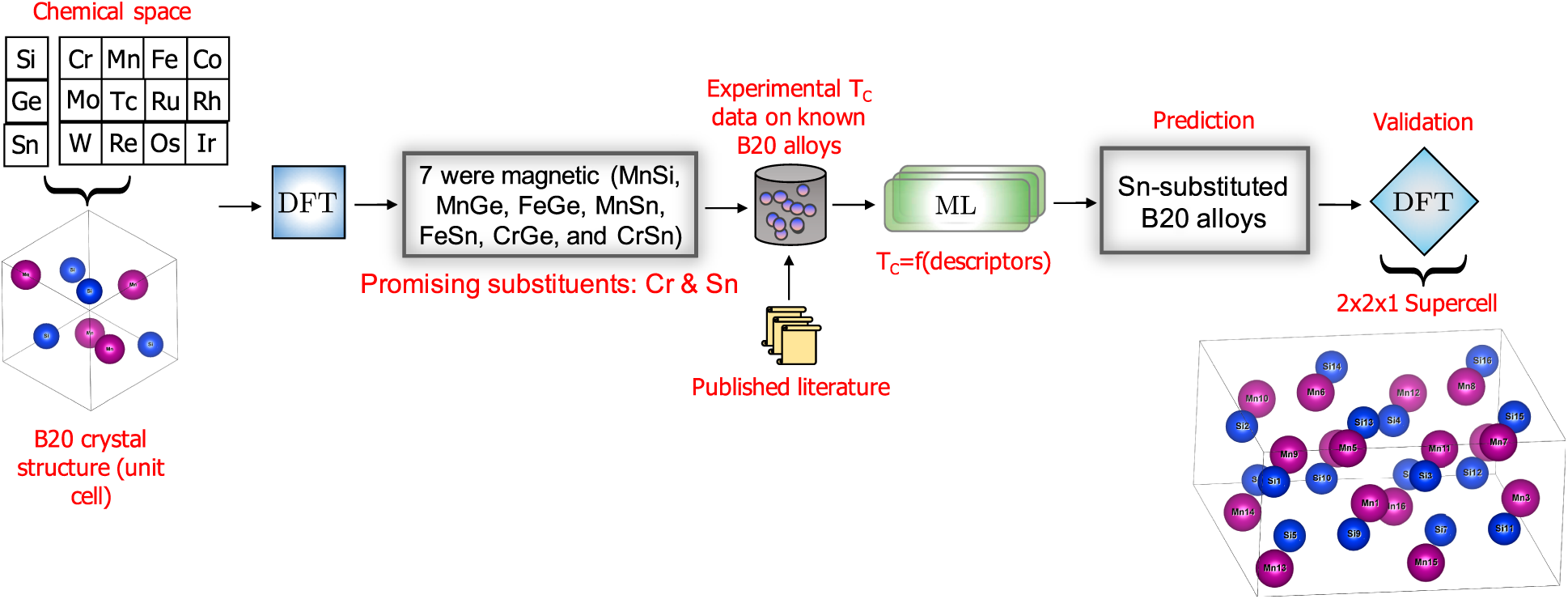
The overarching data-driven strategy is schematically shown in Fig. 2. The data-driven search was initiated by identifying a total of 36 binary compounds in the AB stoichiometry, where A is a transition metal atom and B = Si, Ge, or Sn. All compounds were constrained in the B20 bulk crystal structure (although B20 is not the lowest energy crystal structure for many of the AB compounds explored in this work). Spin-polarized DFT calculations were performed to screen for AB compounds, such that the converged structure is ferromagnetic (see “Methods” for more details), which is a necessary condition for stabilizing a skyrmion phase in the temperature–field phase diagram. The purpose of these initial DFT calculations is not to accurately calculate the atomic magnetic moment of the transition metals in the B20 structure, because DFT is known to overestimate the magnetic moment for some of the B20 compounds (e.g., MnSi [Reference Choi, Lin and Zhu18]). Instead, this step serves as an accelerated screening criterion to downselect promising alloy substituents. The key parameter of interest is the atomic magnetic moment and only those B20 compounds with total magnetization >0.1 Bohr magneton per cell were downselected as candidates from DFT. Out of the 36 compounds that were explored during this initial screening stage, only seven compounds (MnSi, MnGe, MnSn, FeGe, FeSn, CrGe, and CrSn) satisfied this criterion. Three of the seven screened compounds correspond to MnSi, MnGe, and FeGe, which represent the most widely studied chiral magnets in the B20 materials class that also host a skyrmion phase. This gives confidence in the initial screening step. The DFT optimized lattice constant and atomic magnetic moment data of the A-site that contains the transition metal atoms are given in Table I. One of the important outcomes from the initial DFT screening step is the identification of Cr and Sn as potential substituents for tuning the magnetic properties of B20 alloys. The impact of Cr and Sn alloying elements on the T C of B20 alloys have not been explored in the literature. The next key question is then the following: Can we rapidly assess the potential of Cr and Sn when added as a substitutional impurity to known B20 alloys in impacting the T C of the alloy? We choose ML methods to address this question.

Figure 2: The overarching strategy for accelerating the search for new B20 alloy compositions with improved T C is shown. The initial screening step involves running spin-polarized DFT calculations on the chosen chemical space of 36 AB compounds in the constrained bulk B20 crystal structure, where A is the transition metal atom and B is Si, Ge, or Sn. Out of 36 AB compounds, only 7 converged in the magnetic structure, which is a necessary condition for stabilizing a skyrmion phase in B20 compounds. From the seven downselected compounds, two promising alloying elements were identified, namely Cr and Sn, which had not been explored in the literature. ML models were used to establish a relationship between descriptors that represent the compositions of experimentally known B20 and their measured T C values. The trained models were, in turn, used to predict the potential of Cr- and Sn-substituted B20 alloys in the affecting the T C property. ML models identified Sn as a candidate to improve the T C of B20 alloys. The data-driven predictions were validated using DFT calculations, where the magnetic exchange energy was calculated using a supercell approach. Promising and previously unexplored B20 alloy compositions were identified and are recommended for experimental validation.
TABLE I: List of seven AB compounds in the B20 crystal structure with non-zero A-site atomic magnetic moment. The DFT optimized lattice constant (in Å) and the atomic magnetic moment of the transition metal site (in µB) data are also given.

Starting from a training dataset of 18 alloy compositions, we built descriptors based on orbital radius [Reference Waber and Cromer29], effective principal quantum number [Reference Li and Xue30], ratio of orbital radius to effective principal quantum number, and valence electron number [Reference Rabe, Phillips, Villars and Brown31], which are known to capture the chemical trends of solid compounds [Reference Waber and Cromer29, Reference Li and Xue30, Reference Rabe, Phillips, Villars and Brown31, Reference Balachandran, James, Rondinelli and Lookman32, Reference Goldman and Kelton33, Reference Phillips34, Reference Saad, Gao, Ngo, Bobbitt, Chelikowsky and Andreoni35, Reference Zunger36]. The full list of input descriptors is given in Table II. The alloy compositions used for training and testing the ML models are given in Table III. Each B20 chemical composition is represented in terms of the linear combination of the weighted contribution of these elemental descriptors. For example, the A-site valence electron number for Fe0.2Mn0.8Ge is described as 0.2 × VENFe + 0.8 × VENMn. Separate sets of descriptors were constructed to independently represent A- and B-sites (see Table II). Pairwise statistical correlation analysis indicated a strong positive correlation between the following descriptors: the effective principal quantum number (ePQN), A-site d-orbital radii WCd, and their ratio (ePQN/WCd. On the other hand, the valence electron number showed only weak correlation with the d-orbital radii. Therefore, both valence electron number and ratio of atomic orbital radius to the effective principal quantum number were retained to represent the A-site. In contrast, only the ratio of s, p-orbital radii sum to the effective principal quantum number was used to represent the B-site. The valence electron number of all group IV elements is 4 and show no variation with respect to Si, Ge, or Sn. Therefore, this descriptor was not considered for ML. The finalized list of input descriptors for building ML-based regression models is highlighted in bold in Table II. It is important to note that we did not include either J or DM interactions as input descriptors for building the ML models. This is mainly because calculation of J and DM interactions from DFT requires supercells and non-collinear spins, which are computationally challenging even for a simple compound. Furthermore, our composition space also includes solid solutions of B20 alloys (see Table III), and DFT calculation of J and DM interactions for solid solutions is a nontrivial task.
TABLE II: List of descriptors that were considered for the ML work to establish a relationship between T C and chemical compositions of B20 alloys. Descriptors highlighted in bold were used for building the final ensemble of ML models.

TABLE III: Compositions used for training and testing the SVR models. The experimental and ML predicted T C's, along with the uncertainties (σ), are also given. The chemical space includes both line compounds and solid solutions.

An ensemble of 100 ML models was trained based on the support vector regression (SVR) method for predicting the T C as a function of three input descriptors (see "Methods" for more details). Balachandran and co-workers have demonstrated the potential of ensemble-based SVR methods for building reliable ML models from small data [Reference Holleis, Shivaram and Balachandran27, Reference Xue, Balachandran, Hogden, James, Xue and Lookman38, Reference Balachandran, Xue and Lookman39, Reference Xue, Balachandran, Yuan, Hu, Qian, Dougherty and Lookman40, Reference Balachandran, Shearman, James and Lookman41, Reference Balachandran, Xue, James, Hogden, Gubernatis, Lookman, Lookman, Eidenbenz, Alexander and Barnes42]. The trained models were validated using Mn0.75Rh0.25Ge B20 alloy composition as a test case [Reference Sidorov, Petrova, Chtchelkatchev, Magnitskaya, Fomicheva, Salamatin, Nikolaev, Zibrov, Wilhelm, Rogalev and Tsvyashchenko37], which was recently synthesized by Sidorov et al. using a nonequilibrium high-pressure synthesis method. In the family of Mn1−xRhxGe alloys prepared by Sidorov et al., Mn0.75Rh0.25Ge had the highest T C (125 K). The trained ML models predicted the T C value for Mn0.75Rh0.25Ge compound as 105.6 ± 40.2 K. As shown in Fig. 3, the measured T C falls within the error bar of the predicted values, which is encouraging. Note that Mn0.75Rh0.25Ge was not part of the dataset, that was used for training the ML models. This simple, yet powerful, exercise showed that the trained ML models have predictive capabilities and can be used to rapidly predict the T C for previously unexplored B20 alloy compositions.

Figure 3: The performance of trained ML models on the dataset where the measured and ML predicted T C values are shown in x- and y-axis, respectively. The in-sample points are shown as black circles and the test compound (Mn0.75Rh0.25Ge [Reference Sidorov, Petrova, Chtchelkatchev, Magnitskaya, Fomicheva, Salamatin, Nikolaev, Zibrov, Wilhelm, Rogalev and Tsvyashchenko37]) is depicted as magenta diamond. The dashed red line indicates the x = y line, where the measured and predicted values are exactly the same. The error bars indicate the standard deviation from the prediction of an ensemble of 100 ML models.
We specifically focused on the effect of Cr and Sn substitutions on some of the experimentally known B20 alloys, namely MnSi, MnGe and FeGe, to maximize the impact of our predictions and motivate new experiments. On the basis of chemical similarity, we assumed that Cr randomly substitutes the Mn- and Fe-site, whereas Sn randomly substitutes the Si- or Ge-sites. The trained ML models consistently predicted Cr to decrease the T C of FeGe (best in the training set) at a rapid rate, i.e., the ML predicted T C values for Fe0.9Cr0.1Ge and Fe0.8Cr0.2Ge with increasing Cr content were 220 ± 14.4 and 178.4 ± 19.9 K, respectively. On comparing this result with the ML predicted T C for unsubstituted FeGe (261.1 ± 13.2 K), it can be inferred that Cr substitution is unfavorable for improving the T C of FeGe compound. However, Cr substitution increased the T C of MnSi, i.e., the ML predicted T C values for Mn0.9Cr0.1Si and Mn0.8Cr0.2Si were 32.9 ± 9.1 and 44.9 ± 16.4 K, respectively. Furthermore, Cr substitution only had a marginal or no effect on the T C of MnGe (the ML predicted T C values for Mn0.9Cr0.1Ge and Mn0.8Cr0.2Ge were 141.5 ± 36.6 and 141.54.9 ± 45.3 K, respectively). Large ML prediction uncertainties indicate lack of knowledge on the (Mn,Cr)Ge material system.
In contrast, Sn-substituted FeGe alloys showed promise. The ML predicted T C values for FeGe0.9Sn0.1, FeGe0.8Sn0.2, and FeGe0.75Sn0.25 were 259 ± 13.2, 257 ± 13.2, and 255 ± 13.3 K, respectively. Intriguingly, Sn substitution did not have any discernible impact on the T C of FeGe alloys. Unlike the Cr substitution of FeGe that resulted in a sharp TC decrease, the addition of Sn is predicted to only marginally decrease the T C of FeGe compound. This outcome warrants an explanation. It is important to recognize that extrapolation is considered a challenging problem for ML methods, especially when these methods are used without an adaptive learning scheme or iterative feedback loop [Reference Lookman, Balachandran, Xue and Yuan43]. Balfer and Bajorath [Reference Balfer and Bajorath44] showed that when SVR methods are used for building quantitative structure–activity relationships, these methods systematically underestimate the true value of high-potency compounds due to the regularization term in the loss function that balances the trade-off between model complexity and its ability to generalize to an unseen data point. Therefore, our ML prediction for Sn-substituted FeGe needs further attention and cannot be discarded solely on the basis of ML predictions. Similar to Cr substitution, the addition of Sn increased and only had a marginal effect on the T C for MnSi and MnGe, respectively. The ML predicted T C values for MnSi0.9Sn0.1, MnSi0.8Sn0.2, MnGe0.9Sn0.1, and MnGe0.8Sn0.2 were 32.5 ± 8.7, 41.6 ± 9.9, 141.9 ± 28.6, and 140.6 ± 28.4, respectively. The next step involved validating the data-driven predictions of Sn-substituted B20 alloys using DFT calculations.
A new set of DFT calculations were then performed to further understand the predictions from the data-driven ML models (see “Methods” for more details). A Heisenberg Hamiltonian was used to describe the magnetic exchange energy difference in the ferromagnetic ground state at T = 0 K, which can be written as follows [Reference Kübler, William and Sommers45]:
 $$\Delta E\;\infty \;{S^2}Jz\quad ,$$
$$\Delta E\;\infty \;{S^2}Jz\quad ,$$where ΔE is the total energy difference between ferromagnetic and antiferromagnetic spin states, z is the coordination number for (oppositely) ordered moments, S is the local magnetic moment at the atomic sites, and J is the exchange interaction energy between two spins. Thus, Eq. (1) relates ΔE and J. When the value of J is known, one can use the well-known mean field approximation formula to estimate the paramagnetic Curie temperature [Reference Kübler, William and Sommers45, Reference Dhital, DeBeer-Schmitt, Zhang, Xie, Young and DiTusa46]:
 $$T_{\rm{C}}^\Theta \;\infty \;{{{S^2}J} \over {{k_{\rm{B}}}}}\quad ,$$
$$T_{\rm{C}}^\Theta \;\infty \;{{{S^2}J} \over {{k_{\rm{B}}}}}\quad ,$$where k B is the Boltzmann's constant. From Eqs. (1) and (2), we can also use ΔE to indirectly infer about T CΘ or T C. The calculated values of ΔE for MnSi, MnSi0.75Sn0.25, FeGe, and FeGe0.75Sn0.25 using the supercell approach are given in Table IV. The substitution of Sn increases the ΔE for MnSi relative to the pure MnSi compound. Similarly, the ΔE for FeGe0.75Sn0.25 is greater than that for pure FeGe. Thus, the ΔE data from DFT indicate that the substitution of Sn will increase the T C of MnSi and FeGe. This result agrees well with the ML predictions for Sn-substituted MnSi. However, there is a disagreement between ML predicted T C and DFT calculated ΔE for Sn-substituted FeGe. It is unclear if the SVR method likely suffered from the known pitfall of systematic underestimation due to its poor extrapolative capabilities [Reference Balfer and Bajorath44]. On the other hand, the promising prediction of Sn-substituted FeGe from DFT indicates that the FeGe0.75Sn0.25 composition is worthy of experimental investigation.
TABLE IV: The predictions of T C (in K) from trained ML models and the magnetic exchange energy, ΔE (in meV), from DFT, as described by the Heisenberg Hamiltonian for the promising new alloys. Positive values of ΔE suggest that the ferromagnetic spin states are lower in energy than the antiferromagnetic spin states. The experimentally measured T C values for MnSi and FeGe are 30 and 278 K, respectively.

In the literature, Seow and Ziegler [Reference Seow and Ziegler47] suggested a remedial measure to overcome the SVR underprediction problem. This involves synthetically increasing the proportion of the high value points using bootstrapping or oversampling, so that the relative proportion of the high value data points becomes large. Seow and Ziegler demonstrated the efficacy of this approach on an environmental engineering problem. However, this method was not considered for the current study because of the smaller size of the training dataset. The skyrmion dataset is at least thirty times smaller than the environmental engineering problem, which had 638 data points to train the SVR models. Therefore, experimental validation remains one of the promising avenues to test the underprediction problem and provide feedback for ML model improvement.
In addition to the known B20 compounds, Table IV also lists a prediction for one more compound, namely FeSn, in the hypothetical B20 crystal structure. The novelty of this result stems from the fact that this compound has never been synthesized in the B20 crystal structures. Its ML predicted T C of 234.8 ± 14.2 is greater than that of MnGe (which has an experimentally determined T C of 170 K), but is lower than that for FeGe0.75Sn0.25. Another encouraging aspect of bulk FeSn compound is that it has been experimentally synthesized, although in a centrosymmetric B35 crystal structure (space group, P6/mmm). To determine if the B20 structure is energetically close to B35, the total energies for FeSn in both B20 and B35 crystal structures were calculated. The B20 structure was found to be +211.8 meV/atom higher in energy than the B35 crystal structure. Thus, FeSn in B20 structure is predicted to be highly metastable. Similar to FeSn, FeGe has also been synthesized in the B35 crystal structure [Reference Tomiyoshi, Yamamoto and Watanabe48]. Nevertheless, the B20 FeGe is only +17.6 meV/atom higher in energy than the B35 crystal structure. As discussed earlier, nonequilibrium synthesis routes [Reference Zhang, Han, Ge, Du, Jin, Wei, Fan, Zhang, Li and Zhang12] were able to surmount the energy barrier and stabilize FeGe in the B20 crystal structure.
Conclusions
A data-driven computational approach is demonstrated to accelerate the design of novel chiral magnets in the B20 crystal structure with targeted T C property. This work highlights how ML methods can be used to efficiently guide the more computationally intensive physics-based DFT calculations toward promising directions in the composition space. This is especially beneficial when the costs of running the high-fidelity calculations are high. New alloy compositions are identified as promising with likely improved T C values, namely (Cr, Mn)Si, Mn(Si, Sn), and Fe(Ge, Sn), relative to the parent compounds, MnSi and FeGe. These results motivate new experiments to validate the computational predictions and provide feedback for model improvement. One of the major outcomes of this work is the prediction of Fe(Ge, Sn) with improved T C values, which can have key implications in the design of skyrmions for room temperature spintronics applications.
Methods
Density functional theory
DFT calculations were performed within the generalized gradient approximation, as implemented in Quantum ESPRESSO [Reference Giannozzi, Baroni, Bonini, Calandra, Car, Cavazzoni, Ceresoli, Chiarotti, Cococcioni, Dabo, Dal Corso, de Gironcoli, Fabris, Guido, Gebauer, Gerstmann, Gougoussis, Anton, Lazzeri, Martin-Samos, Marzari, Mauri, Mazzarello, Paolini, Pasquarello, Paulatto, Sbraccia, Scandolo, Sclauzero, Seitsonen, Smogunov, Umari and Wentzcovitch49]. The PBEsol exchange–correlation functional [Reference Perdew, Ruzsinszky, Csonka, Vydrov, Scuseria, Constantin, Zhou and Burke50] was used and the core and valence electrons were treated with ultrasoft pseudopotentials [Reference Vanderbilt51]. The Brillouin zone integration used Marzari–Vanderbilt smearing [Reference Marzari, Vanderbilt, De Vita and Payne52], with a smearing width of 0.27 eV and a 12 × 12 × 12 Monkhorst–Pack [Reference Monkhorst and Pack53] k-point mesh centered at Γ. We used 60 Ry plane-wave cutoff for wave functions and 600 Ry kinetic energy cutoff for charge density and potential. The scalar relativistic pseudopotentials were taken from the PSLibrary [Reference Dal Corso54]. The atomic positions and the cell volume were allowed to relax until an energy convergence threshold of 10−8 eV and Hellmann–Feynman forces less than 2 meV/Å were achieved. For the initial accelerated screening step, collinear ferromagnetic spin configurations were imposed on the A-site transition metal atoms, where all spins are constrained to point in the same direction.
The magnetic exchange energy difference (ΔE) was calculated as the total energy difference between the ferromagnetic and the lowest energy antiferromagnetic spin states. Several antiferromagnetic spin configurations were explored and the one with the lowest energy was taken as the most favorable antiferromagnetic spin configuration for the calculation of ΔE. A 2 × 2 × 1 supercell with 32 atoms was considered for the ΔE calculation. In the case of calculations that involved Sn substitution, four Si or Ge atoms were substituted by the Sn atoms. The Sn–Sn bond distances were maintained at 4.639 and 4.715 Å in the supercells of MnSi0.75Sn0.25 and FeGe0.75Sn0.25, respectively.
Machine learning
All ML analyses were formed using the R statistical environment [55]. The SVR was performed using the method implemented in the e1071 package [Reference Meyer, Dimitriadou, Hornik, Weingessel and Leisch56]. More details about the SVR method can be found in the literature [Reference Smola and Schölkopf57, Reference Friedman, Hastie and Tibshirani58]. We used the nonlinear radial basis function (RBF) kernel to establish a quantitative relationship between the input descriptors (see Table II) and experimentally determined T C. Error bars for each prediction were estimated using the bootstrap resampling method [Reference Bradley59, Reference MacKinnon, Lockwood and Williams60]. The hyperparameters for the SVR were optimized using leave-one-out cross-validation method for each bootstrap sample. The trained SVR models are used to predict the T C for unexplored alloy compositions. From 100 SVR models, we get 100 predicted T C values for each composition. The mean (µ) and standard deviations (error bar, σ) are estimated from the 100 SVRRBF models.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1557/jmr.2020.38.
Acknowledgments
This work was financially supported by the Defense Advanced Research Project Agency (DARPA) program on Topological Excitations in Electronics (TEE) under Grant No. D18AP00009. All DFT calculations were performed in the Rivanna high-performance computing cluster maintained by the Advanced Research Computing Service at the University of Virginia.








 Open access
Open access