

The metabolic side-effects of antipsychotics represent a major and unresolved clinical problem facing psychiatry. Clozapine, the most effective antipsychotic for treatment-resistant schizophrenia, Reference Mcilwain, Harrison, Wheeler and Russell1 is strongly associated with weight gain, glucose intolerance and new-onset type 2 diabetes mellitus. Reference Allison, Mentore, Heo, Chandler, Cappelleri and Infante2 More than 20% of patients treated with clozapine experience at least a 10% increase in body weight, Reference Wetterling3 and the rate of new-onset type 2 diabetes mellitus has been reported as 37% at 5 years. Reference Henderson, Cagliero, Gray, Nasrallah, Hayden and Schoenfeld4 In addition to adversely affecting quality of life and adherence, the metabolic adverse effects of clozapine cause increased cardiovascular morbidity and premature death. Reference Newcomer5,Reference Allison, Mackell and McDonnell6 Cardiovascular mortality in schizophrenia is double that of the general population, representing a growing disparity of considerable public health relevance. Reference Saha, Chant, Welham and McGrath7
The current evidence for pharmacological and non-pharmacological interventions for clozapine-associated obesity is limited and indicates only modest benefit as shown in a recent systematic review. Reference Whitney, Procyshyn, Fredrikson and Barr8 Adjunctive metformin appears most promising; however, this is based on two small randomised controlled trials (RCTs) which report modest metabolic improvement and weight regain after treatment cessation. Reference Carrizo, Fernandez, Connell, Sandia, Prieto and Mogollon9,Reference Chen, Huang, Kao, Lin, Kuo and Chiu10 Metformin is also contraindicated in renal impairment and not all patients tolerate metformin.
Although the physiological mechanisms underlying clozapine's metabolic adverse effects are not fully understood, recent preclinical models have suggested glucagon-like-peptide-1 (GLP-1) is a key mediator. Reference Smith, Vickers, Cognard and Shepherd11 GLP-1 is an endogenous peptide synthesised in the intestinal mucosa. Reference Holst12 GLP-1 decreases glucagon secretion and stimulates insulin secretion in a glucose-dependent manner, delays gastric emptying and appears to lower food intake by promoting satiety. Reference Holst12 Clozapine acutely reduces GLP-1 secretion in rodents to cause increased hepatic glucose output and glucagon levels and a preference for high fat and high sugar foods. Reference Smith, Vickers, Cognard and Shepherd11,Reference Smith, Chaussade, Vickers, Jensen and Shepherd13
GLP-1 receptor agonists, including exenatide and liraglutide, normalise glucose tolerance and decrease body weight in rats treated with clozapine and olanzapine, providing mechanistic justification of their therapeutic potential in this context. Reference Smith, Vickers, Cognard and Shepherd11,Reference Lykkegaard, Larsen, Vrang, Bock, Bock and Knudsen14 Additionally, preclinical studies indicate that GLP-1 agonists have effects beyond their glucoregulatory actions including an antipsychotic-like effect as well as procognitive, neuroprotective and cardioprotective effects, which may provide multifactorial benefits for patients with schizophrenia. Reference Dixit, Sharma, Lucot and Elased15,Reference Ryan and Acosta16
GLP-1 agonists have established glucose and body weight lowering properties in diabetic non-psychiatric patients, underlying their successful clinical application as anti-diabetic agents. Reference Shyangdan, Royle, Clar, Sharma and Waugh17 GLP-1 agonists may also improve beta cell mass and function. Reference Gedulin, Nikoulina, Smith, Gedulin, Nielsen and Baron18,Reference Xu, Stoffers, Habener and Bonner-Weir19 Additionally, a growing body of evidence supports that GLP-1 agonists cause clinically significant and sustained weight loss for non-diabetic obese patients and may reduce the prevalence of pre-diabetes. Reference Zhang, Tong, Su, Li, Tang and Huang20
On the basis of this promising preclinical and clinical data and the physiological mechanism of action, GLP-1 agonists may represent a novel approach for treating clozapine-associated obesity and type 2 diabetes mellitus and its underlying pathophysiology. A recent case report provided the first clinical evidence in support of this theory. Reference Ishøy, Knop, Vilsboll, Glenthoj and Ebdrup21 Given the identified need to improve cardiometabolic health among clozapine-treated patients, further clinical research is needed. Two ongoing clinical trials are currently examining GLP-1 agonists for antipsychotic-associated obesity and glucose tolerance in obese, non-diabetic patients. Reference Ishøy, Knop, Broberg, Baandrup, Fagerlund and Jørgensen22,Reference Larsen, Vedtofte, Holst, Oturai, Kjaer and Corell23 However, since clozapine is reserved for treatment-refractory schizophrenia and is associated with greater metabolic disturbances than all other antipsychotics, clinical research investigating strategies specific to this distinct sub-population of clozapine-treated patients is of significant therapeutic interest.
Given the paucity of research in this area, the treatment of clozapine-associated obesity and diabetes with exenatide (CODEX) protocol has been designed as a pilot RCT. The primary aim is to evaluate the feasibility as well as acceptability and tolerability of weekly exenatide for obesity and diabetes in clozapine-treated patients. This will provide guidance regarding the value and design of a large-scale RCT. In addition, we will investigate the preliminary effects of exenatide on clinically important outcomes including glycaemic control and weight.
Method
Study design and setting
The CODEX study is a 24-week, investigator-initiated, open-label, parallel, randomised, controlled pilot trial in 60 obese (body mass index (BMI) ≥30 kg/m2) adults with schizophrenia and on stable treatment with clozapine, with or without poorly controlled diabetes (HbA1c ≥7.5%). With the knowledge that diabetes is a potential confounding factor, the study has been designed with two arms: arm A includes obese patients with inadequately controlled type 2 diabetes mellitus, and arm B includes obese patients without type 2 diabetes mellitus. These two strata of patients have been included since preclinical data have implicated GLP-1 in the pathophysiology of both conditions. Reference Smith, Vickers, Cognard and Shepherd11,Reference Lykkegaard, Larsen, Vrang, Bock, Bock and Knudsen14 Within each arm, patients will be randomised to exenatide (intervention group) or treatment as usual (control group) for 24 weeks. This study will be conducted within a single out-patient mental health service which operates community clozapine clinics: Metro South Addiction and Mental Health Service (MSAMHS), Brisbane, Australia.
Study population
The CODEX trial will recruit obese patients with or without poorly controlled diabetes, who are diagnosed with schizophrenia or schizoaffective disorder and on stable treatment with clozapine. The detailed inclusion and exclusion criteria are presented in the Appendix.
Patient screening and enrolment
Clozapine is a highly specialised drug that requires routine and comprehensive clinical monitoring for safety and efficacy. Therefore, as part of standard care, all patients are required to attend routine (at least four-weekly) appointments at MSAMHS community clinics for monitoring of clinical progression, physical health, blood haematology and metabolic parameters.
Psychiatrists or psychiatry registrars will screen clozapine-treated patients attending MSAMHS clinics for eligibility by reviewing past medical records and recent physical- and laboratory-based assessments. Participants meeting inclusion and exclusion criteria will be invited to participate in the study and the formal consent process will commence. Patients will receive oral and written information about the aims, design, risks and benefits of the trial and will provide written informed consent prior to any trial-related intervention. For participants whose mental impairment interferes with their capacity to adequately consent to the research, consent will be obtained from an adult legal guardian. Consenting participants will be enrolled into the study in either arm A or arm B according to their clinical diagnosis of diabetes. Enrolment will proceed until 15 control participants and 15 intervention participants in each of the two arms have been recruited (total 60 patients). Figure 1 presents a participant flow chart from screening to follow-up.

Fig. 1 Flow chart of the CODEX trial. BMI, body mass index.
Randomisation and masking
Following baseline investigations, participants will be randomised to the intervention (exenatide) or control group (treatment as usual) in a 1:1 ratio using block randomisation. Using the random allocation rule, Reference Schulz and Grimes24 cards denoting allocation will be placed in sealed opaque envelopes in randomly ordered blocks of less than six. Since the study does not incorporate a placebo and involves exenatide administration by a nurse, patients and nurses cannot be masked to the intervention. Carers, data collectors and outcome assessors will not be masked to treatment allocation either.
Although assessments will be conducted in an unmasked fashion, outcomes are primarily objective (physical measurements and laboratory values) and thereby the unblended design is unlikely to bias the data. However, the absence of masking for raters and patients has potential to influence the subjective mental health questionnaire (Brief Psychiatric Rating Scale-Anchored, BPRS-A). We recognise that this may diminish the validity of evidence for this outcome; however, this is appropriate given this is not a primary outcome measure for this proof-of-concept trial. Should data be promising, larger placebo-controlled, masked studies are anticipated.
Treatment protocol
Exenatide group
The intervention group will receive a once-weekly subcutaneous abdominal injection with exenatide 2 mg, in addition to routine clinical care, for 24 weeks. Exenatide will be administered by a trained mental health nurse or a clinical trial nurse. This will maximise adherence to the intervention and will allow accurate, objective measurement of adherence. Dose titration is not required and dose or frequency will not be changed. This dosing regimen is in accordance with current clinical recommendations. 25 To minimise the burden of participation, exenatide will be administered in the patient's home or in the community clinic if dosing coincides with routine clinic appointments.
Supplies of study medication will not be provided to participants; rather, single-dose kits will be collected from the out-patient clinic and administered to the participant by a nurse on a weekly basis. In accordance with good clinical practice, an investigational product accountability record form will be maintained. Exenatide will be sourced, assembled and packaged by the commercial manufacturer.
Control group
Participants in the control group will continue to receive treatment as usual for the 24-week study period. In this study, treatment as usual is defined as individualised combinations of pharmacotherapy, behavioural interventions, rehabilitation and associated clinical services in keeping with Queensland Health Standards of Care.
This study is not placebo-controlled due to ethical concerns about using placebo injection within the context of a pilot study in mental health patients, as well as practical considerations (budget constraints, reduction in recruitment or retention rates). Employing usual care controls avoids unnecessary psychosocial harms while allowing for realistic examination of acceptability and relative retention in intervention groups. Active comparators were also considered premature as the benefit of exenatide in this population has yet to be established.
Concomitant treatment
Details of concomitant medication will be recorded at every trial visit. Owing to their potential to confound the outcome measures, medications that are not permitted include: insulin, corticosteroids or hormone therapy for >10 days and weight-lowering therapy (including pramlintide, sibutramine, orlistat, zonisamide, topiramate or phentermine).
By providing informed consent, the participants consent that their general practitioner (GP) will be informed of their participation in the study. GPs will be requested to communicate with the study endocrinologist about any urgent concerns they have about glycaemic control and need for medication changes. Any change to glycaemic agents will be initiated by the study endocrinologist. Participants with diabetes will be deemed to have deteriorating glycaemic control if their week 12 HbA1c result exceeds the baseline value. For participants who are on maximal sulphonylurea dosage, insulin will be offered. Participants who commence on insulin will be discontinued from the study. Insulin will be initiated by the study endocrinologist and subsequently managed by the participant's GP. Participants who do not accept insulin may continue in the trial. Participants with deteriorating glycaemic control who are not on maximal sulphonylurea dosage will have their dosage optimised and may continue participation in the trial.
For participants in the control arm, initiation of glucose modifying agents (excluding GLP-1 agonists) during the trial will be recorded in the case report form but trial participation may continue. Only control participants who are commenced on a GLP-1 agonist will be discontinued from the trial.
Patients receiving co-treatment with a sulphonylurea and who have lower relative HbA1c are at increased hypoglycaemia risk with weekly exenatide. Reference Paul, Maggs, Klein and Best26 Therefore, for sulphonylurea-treated patients in the intervention group with an HbA1c of 7.5–8.5%, the dose of the sulphonylurea will be halved at study entry. Sulphonylurea dose will be maintained for patients with HbA1c >8.5%. This is in line with standard recommendation from the product information for exenatide extended release.
In this study, dipeptidyl peptidase-4 inhibitor (gliptin) therapy will not occur concurrently with exenatide since these agents both potentiate the actions of endogenous GLP-1. For patients in the intervention arm, gliptin therapy will be ceased just prior to exenatide administration. Control patients may continue receiving gliptin therapy throughout the trial.
There are no restrictions in terms of diet, exercise or ambulation or any other concomitant medication, including psychotropics. Routine care provided by MSAMHS regarding lifestyle recommendations and management of coexisting illnesses will be continued as usual for all participants.
Discontinuation and post-trial care
The investigator may discontinue patients in the study in the event of adverse events, protocol violations or non-adherence, inter-current illness or administrative reasons. All participants and surrogate decision makers also have the right to withdraw consent at any time without affecting current or future care. Withdrawal (and the primary reason) will be recorded in the case report form and, where possible, follow-up data collection will be continued. Participants who are discontinued will not be replaced and are not permitted to re-enter the study.
As discussed above, participants who continue to have poor glycaemic control on maximal sulphonylurea dosage will be offered insulin. Only participants who commence insulin will be discontinued from the study.
There are no restrictions on participants in the post-study period and participants will continue to receive ongoing treatment from the mental health service.
Outcomes
Primary outcome
This is to assess the feasibility of using weekly exenatide for the treatment of clozapine-associated obesity and diabetes. Key indicators are the number of eligible participants, the recruitment and retention rates as well as the acceptability and tolerability of the intervention, as indicated by study drop-out rates, adverse event rates and patient-reported questionnaire data for patient satisfaction.
We will also collect preliminary clinical outcome data. For patients with diabetes (arm A), the primary clinical outcome is the change in glycaemic control after 24 weeks of exenatide, as measured by change in HbA1c, and the proportion of patients with reduction in HbA1c of >0.5% from baseline. In arm B, the primary outcome is the change in weight and proportion of patients with >5% weight change from baseline to week 24. These clinical outcome measures are clinically meaningful: HbA1c is the gold standard measure of glycaemic control and a strong predictor of diabetes morbidity, 27,Reference Turner, Holman, Stratton and Cull28 whereas weight reduction of 5% results in clinically relevant reductions in morbidity and risk of early mortality. 29
Obtaining preliminary data on the magnitude and direction of clinically relevant effects will inform the value and design of a further study and provide data to appropriately power the trial.
Secondary outcome
Secondary clinical end-points include changes in metabolic markers (lipid profile, blood pressure and fasting glucose), symptoms of psychosis as estimated by the BPRS-A, insulin sensitivity as determined by homeostatic model assessment (for arm B) and weight loss (for arm A) from baseline to week 24.
Trial visits and assessments
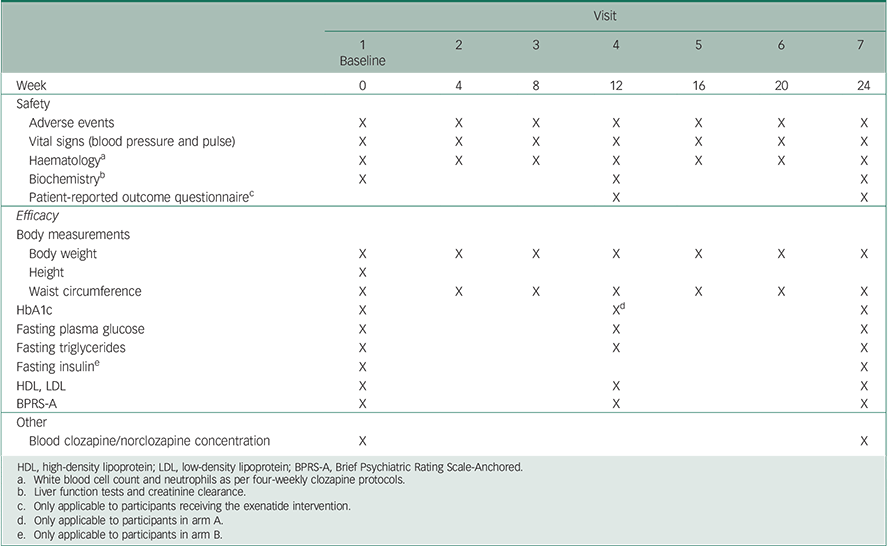
To improve retention and limit the participant burden, all assessments will be conducted during routine care visits within the community clinic. As per clinic protocol, all patients will attend the clinic at four-weekly intervals (from baseline to week 24) for outcome assessments including: blood sampling for metabolic and haematological investigations, physical health measurements (weight, vital signs and waist circumference), evaluation of psychopathology, side-effects and treatment satisfaction ratings. The detailed outcome measures and schedule for trial visits is presented in Table 1.
Table 1 Summary of visit schedule and assessed parameters at each time point
| Visit | |||||||
|---|---|---|---|---|---|---|---|
| 1 Baseline |
2 | 3 | 4 | 5 | 6 | 7 | |
| Week | 0 | 4 | 8 | 12 | 16 | 20 | 24 |
| Safety | |||||||
| Adverse events | X | X | X | X | X | X | X |
| Vital signs (blood pressure and pulse) | X | X | X | X | X | X | X |
| Haematology a | X | X | X | X | X | X | X |
| Biochemistry b | X | X | X | ||||
| Patient-reported outcome questionnaire c | X | X | |||||
| Efficacy | |||||||
| Body measurements | |||||||
| Body weight | X | X | X | X | X | X | X |
| Height | X | ||||||
| Waist circumference | X | X | X | X | X | X | X |
| HbA1c | X | X d | X | ||||
| Fasting plasma glucose | X | X | X | ||||
| Fasting triglycerides | X | X | X | ||||
| Fasting insulin e | X | X | |||||
| HDL, LDL | X | X | X | ||||
| BPRS-A | X | X | X | ||||
| Other | |||||||
| Blood clozapine/norclozapine concentration | X | X | |||||
HDL, high-density lipoprotein; LDL, low-density lipoprotein; BPRS-A, Brief Psychiatric Rating Scale-Anchored.
a White blood cell count and neutrophils as per four-weekly clozapine protocols.
b Liver function tests and creatinine clearance.
c Only applicable to participants receiving the exenatide intervention.
d Only applicable to participants in arm A.
e Only applicable to participants in arm B.
Anthropometric parameters
At each clinic visit, weight will be recorded to the nearest 0.1 kg using calibrated scales. Height without shoes will be recorded at screening. Waist circumference will be measured in the horizontal plane to the nearest 0.5 cm using a non-stretchable measuring tape placed around the abdomen at the level of the top of the hip bone.
Blood analysis
In accordance with clinic monitoring guidelines, 30 continued safety of therapy and adherence to clozapine will be ensured through monthly analysis of blood haematology (full blood count, white cell count and neutrophils), liver function tests (LFTs), creatinine clearance and clozapine/norclozapine levels. Fasting glucose, HbA1c, triglycerides, fasting insulin, high-density lipoprotein (HDL), low-density lipoprotein (LDL) and total cholesterol will be quantified for evaluation of the participant's metabolic status. During the entire study, approximately 60–65 mL of blood will be drawn per participant.
Questionnaires
The BPRS-A is a widely used questionnaire that provides a responsive, valid and reliable measure of positive, negative, and affective symptoms in psychotic disorders. Reference Lachar, Bailley, Rhoades, Espadas, Aponte and Cowan31 It is a brief 18-item instrument rated on a 0- to 7-point scale. The BPRS-A will be used to assess changes in psychosis symptoms.
A patient-reported outcome questionnaire has been developed for this study to measure patient satisfaction and perception of the value and acceptability of exenatide. This will aid in informing the feasibility of weekly exenatide in this population. This instrument has been modelled from a validated patient-reported outcome questionnaire. Reference Atkinson, Sinha, Hass, Colman, Kumar and Brod32 It consists of eight items, rated on a 5-point scale, and investigates two domains: satisfaction and usage and self-reported adverse event frequency. Questionnaires will be administered in a structured qualitative interview by a trained psychiatrist or psychiatry registrar during routine consultation within clozapine clinics.
Statistical methods
Sample size
There is no published data on the outcomes of interest in this patient population to calculate sample size. Due to the exploratory nature of this pilot study the sample size is therefore based on the pragmatic limits of recruitment and patient flow and the necessities for examining feasibility. There are approximately 520 patients attending clozapine clinics at MSAMHS. Preliminary data indicate that approximately 56.7% of clinic attenders have a BMI ≥30 kg/m2 and 26.7% have type 2 diabetes mellitus, of which 37.5% are poorly controlled (HbA1c ≥7.5%). A 40% study refusal rate is anticipated. Therefore, we plan to randomise a total of 30 obese participants with poorly controlled diabetes and 30 obese participants without diabetes.
This number is consistent with published pilot study guidelines which consider samples sizes of 12 per group Reference Julious33 or 30 overall Reference Lancaster, Dodd and Williamson34 to be sufficient for informing feasibility, acceptability and estimates of effect size and variance for sample size calculations in larger trials.
Data analysis
Efficacy outcomes will be assessed according to standard intention-to-treat analytic procedures consisting of all randomised participants in the group to which they were originally allocated. Missing data will be imputed using the last-observation carried forward method. Per protocol analysis will also be performed for the primary analysis. Safety end-points will be analysed in the per protocol population. Data will be analysed as changes from baseline to end of study and for study arms separately.
Continuous data will be summarised using descriptive statistics (mean, median, standard deviation or range as appropriate to the distribution of the data) and categorical end-points will be expressed using counts and percentages. Unless otherwise stated, comparison of data between intervention and control groups will be analysed using independent sample t-tests (for continuous outcomes) and χ 2-tests (for categorical data). Change in scores will also be compared using paired t-tests (continuous) and McNemar's tests (categorical data). Multivariable linear regression analysis will be performed to evaluate the correlation between baseline characteristics and outcome parameters.
All data will be analysed using an SPSS software version 22 (Chicago, Illinois, USA) with significance set at P<0.05. As no formal power calculations have been performed, statistical results should be considered preliminary and interpreted with caution. Reference Lancaster, Dodd and Williamson34
Adverse events and data monitoring
We anticipate that most adverse events will be non-severe and expected treatment-related toxicities. Common adverse events associated with exenatide are mild-to-moderate gastrointestinal disturbances including diarrhoea, vomiting and nausea occurring in up to 20% of patients and typically resolving within 4–8 weeks. Reference Harris and McCarty35 Less frequently, patients describe headache, fatigue, reflux, dizziness and transient injection site redness, itchiness or haematoma. 25 These possible risks are outlined in the participant information sheet, and the likely frequency, severity and duration of side-effects will be discussed as part of the consent process. Hypoglycaemia as monotherapy and in combination with metformin is rare due to the glucose-dependent insulinotropic effects. Reference Shyangdan, Royle, Clar, Sharma and Waugh17
Adverse events are defined and reported to regulatory bodies in accordance with local legislative requirements and good clinical practice guidelines. Investigators will assess all adverse events for seriousness, causality, intensity and expectedness. At each clinic visit, participants will be asked two standardised questions addressing the occurrence of any adverse events and secondly, about specific hypoglycaemic symptoms. Self-monitoring of blood glucose is not feasible among this population; however, all recorded plasma glucose values ≤3.9 mmol/L and self-reported hypoglycaemic symptoms will be considered a hypoglycaemic episode and recorded as an adverse event. The study team will also liaise appropriately with the patient's GP to identify and record details of hypoglycaemic episodes or symptoms consistent with hypoglycaemia and any other reported adverse events.
To facilitate monitoring of the safety, efficacy and integrity of clinical data, this study is subject to periodic audit by the University of Queensland and the Metro South Human Research Ethics Committee under their remit as the reviewing ethical boards. Additionally, an independent psychiatrist will review all adverse events and advise on the continued safety of the trial.
Discussion
Although little research has been conducted in this area to date, several lines of preclinical and clinical evidence converge towards GLP-1-based therapies as a potentially new avenue for treating clozapine-associated metabolic disorders. Repurposing this pre-existing diabetes treatment may cost-effectively reduce cardiovascular morbidity and mortality in these patients with a cost saving to clinical services.
To our knowledge, this is the first trial focusing specifically on clozapine-treated patients to investigate a GLP-1 agonist for glycaemic control and weight loss in both diabetic and obese patients. This study addresses an area of unmet clinical need since there is poor adherence to behavioural advice in this population and the quality and extent of evidence for pharmacological interventions is limited. Reference Whitney, Procyshyn, Fredrikson and Barr8 This pilot study is an important precursor to an appropriately powered multicentre trial, providing the means to evaluate technical aspects of this novel treatment approach (including recruitment and retention rates, economic viability and resource management). Reference Lancaster, Dodd and Williamson34 It also serves as a platform for generating preliminary data for better understanding of whether GLP-1 is a key target for clozapine-associated metabolic disorders in humans. If successful, this programme of research will contribute to evidenced-based strategies to support the clinical management of the severe metabolic side-effects of clozapine in mental health patients.
The need for regular subcutaneous injections with GLP-1-based therapies may represent a barrier to their use in psychiatric populations. We have attempted to minimise this participant burden and improve practicality by an extended release (once weekly) formulation of exenatide over other available twice-daily or daily GLP-1 agonists. Also supporting the choice of this regimen, exenatide weekly has shown greater adherence and treatment satisfaction over other GLP-1 agents, in the context of a currently favourable safety profile. Reference Johnston, Nguyen, Felber, Cappell, Nelson and Chu36,Reference Best, Boye, Rubin, Cao, Kim and Peyrot37 Data from this pilot trial will provide important insight into the challenges and determinants of protocol adherence and acceptability of weekly subcutaneous injections among this patient population to adequately design and power subsequent large RCTs.
Weekly dosing with exenatide exceeds minimally effective therapeutic plasma concentrations after 2 weeks, with most clinical benefit in glycaemic control seen within 12 weeks and progressive weight loss observed within 15 weeks. Reference Fineman, Flanagan, Taylor, Aisporna, Shen and Mace38,Reference Drucker, Buse, Taylor, Kendall, Trautmann and Zhuang39 Therefore, the planned trial duration is sufficient to evaluate exenatide's effects on weight and metabolic status. Although the long-term effects of exenatide or the durability of response cannot be extrapolated from this study, it will be valuable to investigate in future trials after the efficacy of exenatide has been demonstrated.
In conclusion, this pilot, open-label RCT will evaluate whether weekly exenatide as a novel therapy is acceptable to clozapine-treated patients, likely to be effective and if it is feasible to offer in this context, providing a clinical foundation for future larger scale trials.
Funding
This study is an investigator-initiated trial, funded by competitive grants from the Princess Alexandra Hospital Research Support Scheme and the Rebecca L. Cooper Foundation. The funding sources had no role in the design, conduct or data analysis of the present study. The University of Queensland is the research sponsor and will be responsible for monitoring and indemnifying the trial. There are no stipulations on publication in place by any party.
Acknowledgements
We would like to acknowledge Dr Balaji Motamari (Director Psychosis ACU, MSAMHS) for acting as the independent data safety monitor. We would also like to acknowledge Ms Lynette Loy, Director of Pharmacy, PAH for her assistance.
Appendix
Inclusion criteria
(1) Provision of informed consent prior to any study-specific procedures
(2) Male or female aged 18–64 years
(3) Clinical diagnosis of schizophrenia or schizoaffective disorder
(4) On oral clozapine for at least 18 weeks
(5) Stable body weight (defined as less than 5 kg change in weight over the past 3 months before inclusion)
For arm A
(a) Diagnosis of type 2 diabetes mellitus
(b) Current and stable therapeutic doses of oral glucose lowering agents for 3 months prior to recruitment
(c) HbA1c ≥7.5%
(d) BMI ≥30 kg/m2 and <45 kg/m2
For arm B
(a) BMI ≥30 kg/m2 and <45 kg/m2
Exclusion criteria
(1) Pregnancy (screened by urine human chorionic gonadotropin), lactation or no acceptance to use of effective contraception during the intervention period
(2) Severe gastrointestinal disease including gastroparesis and dumping syndrome
(3) Severe renal impairment (creatinine clearance <30 mL/min) or patients with end-stage renal disease receiving dialysis
(4) Allergy/hypersensitivity to exenatide or the investigational product
(5) Obesity induced by other endocrinological disorder (e.g. Cushing's syndrome)
(6) Participants treated with corticosteroids or other hormone therapy (except oestrogens or thyroxine) for more than 10 days
(7) Current use of any weight-lowering therapy including: pramlintide, sibutramine, orlistat, zonisamide, topiramate or phentermine
(8) Uncontrolled hypertension (systolic blood pressure ≥160 mmHg and/or diastolic blood pressure ≥100 mmHg)
(9) Previous surgical treatment of obesity
(10) History of thyroid adenoma or carcinoma
(11) Untreated or uncontrolled hypo/hyperthyroidism (defined as thyroid-stimulating hormone >6 mIU/L or <0.4 mIU/L)
(12) Acute or chronic pancreatitis or high risk of pancreatitis (including a history of pancreatitis, gallstones, alcoholism and severe hypertriglyceridaemia)
(13) Concurrent use of insulin
(14) Any concomitant disease or condition that according to the investigator's assessment makes the patients unsuitable for trial participation
(15) Refusal to have regular injections
(16) For arm B: diagnosis of type 1 or type 2 diabetes mellitus



 Open access
Open access
eLetters
No eLetters have been published for this article.