



Hypertrophic cardiomyopathy is a cardiac condition characterised by diastolic dysfunction and systolic outflow obstruction of the left ventricle. It is inherited in an autosomal dominant manner with beta-myosin heavy chain (MYH7) and myosin binding protein C (MYBPC3) being the most common sites of mutation.Reference Richard, Charron and Carrier1 Although hypertrophic cardiomyopathy has a 1/500 reported prevalence, compound and double mutations for this condition are infrequent.Reference Maron, Gardin, Flack, Gidding, Kurosaki and Bild2,Reference Ingles, Doolan, Chiu, Seidman, Seidman and Semsarian3
We report a case of homozygous MYBPC3 mutations in a 2-year-old presenting with aborted sudden cardiac death and a severe form of hypertrophic cardiomyopathy.
Case report
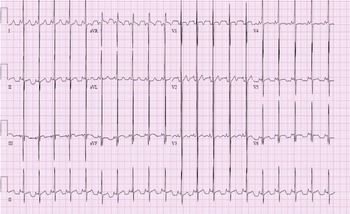
A 2-year-old presented to our paediatric ICU as a transfer from an outside emergency department after an episode of aborted sudden cardiac death. Approximately 3 hours before arrival, she collapsed at home after clutching her chest. Her father began CPR and police arrived with an AED. The AED interpreted the rhythm as ‘shockable’, a single shock was delivered, and rhythm converted to sinus tachycardia. 12-lead electrocardiogram (ECG), obtained in our paediatric ICU, was significant for sinus rhythm, biventricular hypertrophy, and non-specific ST and T wave abnormality.
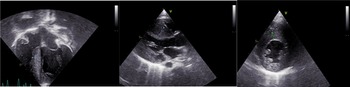
Echocardiography was significant for left ventricular hypertrophy without evidence of obstruction. The interventricular septum measured 1.1 cm by direct measurement with a z-score of 4.02 by Boston data.
The parents indicated there was no family history of sudden cardiac death, CHDs, or hypertrophic cardiomyopathy. Propranolol was initiated to decrease the risk of arrhythmias and increase ventricular fill time. She was then transferred to an institution with paediatric cardiovascular surgery for epicardial implantable cardioverter defibrillator placement (Fig 1).

Figure 1. Electrocardiography (ECG) on arrival to PICU. There are high voltage QRS complexes in the left precordial leads without T wave inversion indicating left ventricular hypertrophy. Prominent mid-precordial voltage indicates possible biventricular hypertrophy. There are also nonspecific ST and T wave abnormalities.
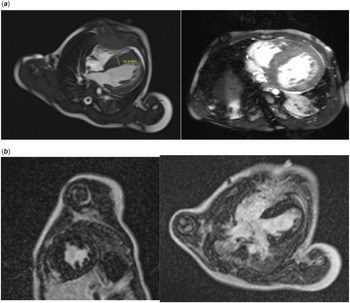
A cardiovascular magnetic resonance with gadolinium contrast was obtained prior to implantable cardioverter defibrillator placement, which was significant for severe septal hypertrophy, mild left ventricular free wall hypertrophy, and increased trabeculation of the free wall and apex (Fig 2). The myocardium of the apex was noted to be thin. The late gadolinium enhancement images showed severe fibrosis with scattered, delayed hyperenhancement of the mid-ventricular septum. A max non-compacted to compacted ratio of 2 was noted, which did not meet criteria for left ventricle non-compaction. The interventricular septum was measured up to 17 mm on left ventricular outflow tract images. Short axis images revealed a septum measurement of 13–14 mm (z-score +18, Boston) and a left ventricle free wall measurement of 6–7 mm (z-score +2 to 3.8, Boston). There was no evidence of left ventricular outflow obstruction. The left ventricle ejection fraction was 57%.

Figure 2. Echocardiographic images showing severe hypertrophic cardiomyopathy. The interventricular septum measures 1.2 cm, 1.8 cm, and 1.0 cm in images 1, 2, and 3 respectively.
A dual chamber epicardial implantable cardioverter defibrillator implantation was placed 4 days after the aborted sudden cardiac death. 4 days after implantable cardioverter defibrillator placement, propranolol was discontinued due to worsening function with a left ventricle ejection fraction of 30% noted on repeat echocardiography. Cardiac catheterisation, without biopsy, was completed due to decreased cardiac function. Catheterisation revealed biventricular diastolic dysfunction (RVEDP 16 mmHg, LVEDP 16 mmHG), a mildly elevated pulmonary artery pressure (24–25 mmHg), normal cardiac output, vascular resistance, and coronary vasculature origins. However, a myocardial bridge was noted in the proximal left anterior descending artery.
Treatment with amiodarone was initiated 4 days after cardiac catheterisation. Some improved cardiac function (LVEF 40%) was noted following the discontinuation of propranolol (Fig 3a and b). A transplant evaluation was completed. However, listing for transplant was deferred initially due to being clinically stable at that time. Enalapril was considered to reduce afterload but deferred due to increased BUN and poor oral intake.

Figure 3. Cardiac MRI images significant for severe septal hypertrophy and increased trabeculation. The For Peer Review interventricular septum measures up to 16.9 mm.
After discharge, genetic testing revealed a homozygous mutation MYBPC3 c. 1504C>T (p. Arg502Trp), a heterozygous mutation of uncertain significance, NM_007078.3:c2092G>A (p. Ala698Thr), and a benign heterozygous mutation, c.2065G>A (p. Glu689Lys). Both parents possessed MYBPC3 mutations. She was monitored closely with amiodarone and aspirin due to decreased cardiac function. Her neurologic status eventually resolved to baseline after cardiac arrest.
Approximately 9 months after the initial cardiac arrest, one run of ventricular tachycardia occurred, which was successfully defibrillated via implantable cardioverter defibrillator despite amiodarone therapy. Propranolol (0.29 mg/kg/dose every 8 hours) was initiated at that time, her clinical status was monitored closely, and she was listed for transplant. Successful heart transplantation took place about 3 months after the second cardiac arrest with successful defibrillation.
Discussion
Hypertrophic cardiomyopathy has a reported prevalence of 1/500. The frequency of compound, double, and triple mutations have attempted to be described with studies reporting an upwards of 6% of patients with hypertrophic cardiomyopathy having more than one mutation.Reference Maron, Gardin, Flack, Gidding, Kurosaki and Bild2–Reference Van Driest, Vasile and Ommen5 One study reported a prevalence of 0.8% of hypertrophic cardiomyopathy patients with triple mutationsReference Girolami, Ho and Semsarian4. There are very few reports describing homozygous mutations. One study reported three patients having homozygous mutations out of 197 patients with hypertrophic cardiomyopathy.Reference Richard, Charron and Carrier1,Reference Xin, Puffenberger, Tumbush, Bockoven and Wang6–Reference Zahka, Kalidas and Simpson8
Although MYBPC3 is one of the most common mutations seen in hypertrophic cardiomyopathy, only a few cases of homozygous MYBPC3 mutations have been reported. In these accounts, patients are described as having severe hypertrophic cardiomyopathy with symptoms occurring in infancy resulting in early death or heart transplant.Reference Xin, Puffenberger, Tumbush, Bockoven and Wang6–Reference Kelly and Semsarian9
Two studies described homozygous MYBPC3 mutations among an Amish community.Reference Xin, Puffenberger, Tumbush, Bockoven and Wang6,Reference Zahka, Kalidas and Simpson8 Affected children presented with symptoms of heart failure shortly after birth and resulted in either death before 1 year of age or heart transplant despite treatment.Reference Xin, Puffenberger, Tumbush, Bockoven and Wang6–Reference Zahka, Kalidas and Simpson8 All children described from this Amish community had splice site mutations in intron 30 (3330 + 2T>G), which were traced to an ancestral founder mutation.Reference Zahka, Kalidas and Simpson8
There is pronounced phenotypic variation between individuals with hypertrophic cardiomyopathy, even with the same mutation or within the same family, making disease course difficult to predict.Reference Richard, Charron and Carrier1,Reference Kelly and Semsarian9–Reference Maron10 Multiple mutations for hypertrophic cardiomyopathy within the same individual could have significant implications in genetic counselling and patient management. Studies suggest that multiple hypertrophic cardiomyopathy mutations tend to confer a worse prognosis with a more severe phenotype.Reference Richard, Charron and Carrier1,Reference Ingles, Doolan, Chiu, Seidman, Seidman and Semsarian3–Reference Girolami, Ho and Semsarian4,Reference Kelly and Semsarian9 Patients with multiple mutations tend to demonstrate increased left ventricular hypertrophy, incidence of sudden cardiac death, risk of end-stage progression, and ventricular arrhythmias.Reference Ingles, Doolan, Chiu, Seidman, Seidman and Semsarian3–Reference Girolami, Ho and Semsarian4
Our patient having a significantly worse disease course, and much earlier onset, than both parents supports this notion. Cardiovascular magnetic resonance showed advanced fibrosis at an early age, further supporting more severe pathology due to homozygosity.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of interest
None.

 Open access
Open access