


INTRODUCTION
West Nile virus (WNV), once considered a pathogen of the Old World, has re-emerged in the past 20 years in the Western Hemisphere, Eastern Europe, and Israel, causing considerable morbidity and mortality [Reference Gubler1–Reference Weinberger5]. Most WNV human infections are subclinical and ~20% of infected patients will present with a mild form of the disease entitled West Nile fever (WNF). However, less than 1% of those infected will suffer from a severe and potentially life-threatening disease designated West Nile neuroinvasive disease (WNND) characterized by meningitis (West Nile meningitis; WNM) and/or encephalitis (West Nile encephalitis; WNE) with or without paralysis (WNE/P) [Reference Murray, Walker and Gould6, Reference Petersen and Hayes7].
The recent spread of WNV in the New World has been attributed to several factors, among which are the emergence of a WNV strain with greater epidemic potential and virulence in birds, humans and horses [Reference Giladi8–Reference Zeller and Schuffenecker10], a hybrid mosquito with greater capabilities of transmitting the virus to humans [Reference Couzin11], and global warming that may have accelerated mosquito development as well as viral infection, dissemination, and transmission through increased virus replication within the mosquito [Reference Kilpatrick12]. Yet, some countries in Asia and Europe did not show any significant increase in data reporting of WNV human disease. This discrepancy has been attributed to variable influence of global warming on certain geographical areas in Europe [Reference Paz13], others suggested that the migratory route of birds, the amplifying hosts of the virus, between Africa, Europe, and the USA may have shaped patterns of data reporting to certain countries [Reference Reed14]. Finally, variable genetic susceptibility to WNV infection in the affected human populations may have impacted its global epidemiology [Reference Samuel15].
In recent years several studies have addressed host genetic predisposition to WNV [Reference Yakub16–Reference Loeb23], these human association studies were exclusively conducted in the USA and investigated several factors that could influence the risk for initial infection with WNV and the likelihood of neuroinvasive disease. Two single nucleotide polymorphisms (SNPs) in the oligoadenylate synthetase (OAS) gene cluster, SNPs rs3213545 and rs10774671, were found to be associated with an increased risk for initial infection with WNV [Reference Yakub16, Reference Lim19]. One study showed that two SNPs (rs2304207, rs7280422) in two genes (IRF3 and MX1, respectively) in the interferon pathway were associated with an increased risk for symptomatic disease and another SNP (rs3413772) in OAS1 gene was associated with an increased risk for neuroinvasive disease [Reference Bigham21]. An increased risk for WNV neuroinvasive disease has also been associated with SNPs in the following genes rs2066786 in RFC1 (replication factor C1), rs2298771 in SCN1A (sodium channel, neuronal type 1α subunit), and rs25651 in ANPEP (alanyl aminopeptidase) [Reference Loeb22]. In addition, homozygosity for CCR5Δ32, a non-functional variant of chemokine receptor CCR5, has been initially suggested to be associated with an increased risk for symptomatic infection [Reference Glass17, Reference Lim18]; however, another study failed to replicate these findings [Reference Lim19].
The epidemiology of WNV infection in Israel is rather intriguing both in the regional and national aspects. Regionally, several reports from the Middle East have shown that the seroprevalence of WNV ranged from 1·3% to 26% of the populations [Reference Hurlbut24–Reference Chinikar31]. However, an outbreak of WNV neuroinvasive disease has been reported only in Israel. During July to September 2000 a WNF and WNND outbreak erupted in Israel causing 417 confirmed cases with 35 deaths [Reference Weinberger5], since then disease activity decreased to some extent [Reference Green32]. Locally, nearly 94% of all the confirmed cases to date of WNF/WNND in Israel were in Jews (E. Anis, Ministry of Health, personal communication) despite the fact that nearly 20% of the Israeli population are Arabs. Data from our region showed that nearly 92% of symptomatic WNV disease occurred in Ashkenazi Jews (N. Bisharat, unpublished data). These findings could suggest that Ashkenazi Jews may have a genetic predisposition for symptomatic WNV infection.
Allele frequencies of CCR5Δ32 in Ashkenazi Jews have been estimated to be significantly higher than for Sephardic Jews and Arabs [Reference Lucotte and Smets33, Reference Kantor and Gershoni34]. We hypothesized that the genetic predisposition of Jews for symptomatic WNV infection could be associated to homozygosity of CCR5Δ32. In the present case-control association study we searched for genetic loci (CCR5Δ32 and several SNPs) that could affect susceptibility to WNV infection in a specific ethnic population.
MATERIALS AND METHODS
Study populations
We reviewed all patients' records who were admitted during the years 1999–2012 in Emek Medical Centre, Afula, with a primary diagnosis of West Nile Fever (WNF), and/or West Nile meningitis (WNM), and/or West Nile encephalitis (WNE), and/or West Nile paralysis (WNP). Fifty-nine patients were admitted during the study period. Five patients had died, the rest (n = 54) were located and were asked to participate in the study, seven declined and the rest (n = 47) agreed to participate in the study. Forty-four patients were Jews and three were Arabs. From the 44 Jewish patients, 39 were Ashkenazi Jews and these were recruited for the case-control study. Informed consent was obtained from the patients or their guardians. A diagnosis of acute WNV infection was based on clinical criteria for WNV disease (WNF, WNM, and WNE/P) combined with a WNV-specific IgM antibody in cerebrospinal specimens with or without positive WNV IgM from blood serum. A control group of Ashkenazi Jews, WNV IgG negative, was assembled from hospital staff and elderly residents from the regional district. Plasma and serum specimens were tested for WNV IgM and WNV IgG using enzyme-linked immunosorbent assay (ELISA) kits (Focus Diagnostics Inc., USA).
DNA extraction and genotyping
DNA was extracted from a 2 ml venous blood sample from each individual participating in the study. Genomic DNA was extracted from blood by use of the Flexi Gene DNA kit (Qiagen, USA). The genomic DNA was screened for several genetic loci (Table 1) that have been previously investigated in patients with WNV infection. Testing for frequency of the variant CCR5Δ32 allele was performed as previously described [Reference Eugen-Olsen35]. Briefly, 2 μl genomic DNA was amplified by PCR using primers flanking the site of a 32 base pair (bp) deletion: 5'-CAATGTGTCAACTCTTGACAGG-3' and 5'-ACC TGCATAGCTTGGTCCAACC-3'. These primers amplify a 547 bp fragment on homozygous wild-type DNA, two fragments of 547 bp and 515 bp on heterozygous CCR5Δ32 DNA and one fragment of 515 bp on homozygous deletion. Genomic DNA was amplified by 35 cycles at 94°C for 30 s, 55°C for 30 s and 72 °C for 30 s. Amplified products were separated on 2% agarose gel.
Table 1. Genetic variants investigated in the study

SNP, Single nucleotide polymorphism; n.a., not applicable.
Statistical analysis
The association of SNPs with WNV infection was tested at the allele level and at the genotype level. The associations were tested using χ 2 test (or Fisher's exact test whenever the χ 2 test was not considered valid) with two-sided test. The minor allele was defined based on the allele frequencies in the dataset; odds ratios for the minor allele were calculated using a genotypic model (DD vs. Dd vs. dd), a dominant model (DD and Dd vs. dd), and a recessive model (DD vs. Dd and dd). Genotype and allele frequencies in cases and controls were analysed for associations by use of χ 2 test on 2 × 2 and 2 × 3 contingency tables. Univariate and multivariate analyses were performed using logistic regression for the probability of having WNV infection. Statistical analysis was performed using SAS v. 9.2 software (SAS Institute Inc., USA).
Ethical statement
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
The study was approved by the local ethics committee at Emek Medical Centre.
RESULTS
Characteristics of cases and controls
Of 223 individuals (hospital staff and elderly residents) asked to participate in the study, 50 were found to be WNV IgG positive (22·4%). The rest (n = 173) were WNV IgG negative, from whom 61 Ashkenazi Jews were recruited and served as controls. The mean age of cases and controls was 66 and 64 years, respectively. Eighty-five percent of cases suffered from WNE/P, 13% suffered from WNM, and the rest were diagnosed as WNF (Table 2).
Table 2. Characteristics of case patients and controls enrolled in the study
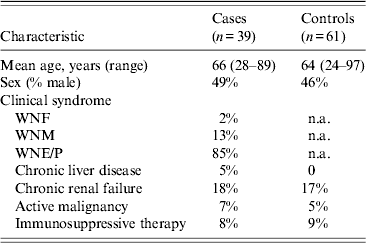
WNF, West Nile fever; WNM, West Nile meningitis; WNE/P, West Nile encephalitis with or without West Nile paralysis; n.a., not applicable.
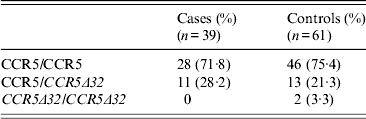
Heterozygozity for the variant allele CCR5Δ32 was 28% in cases and 22% in controls (P = 0·2). None of the case patients was homozygous for the variant allele, compared to 3·3% of controls (Table 3). In order to exclude any association between the CCR5 gene and WNV infection we sequenced the coding sequence (exon 3) of the CCR5 gene (1059 bp, 352 amino acids) in all patients and controls, the analysis failed to show any genetic variants significantly associated WNV infection status.
Table 3. CCR5 genotypes in cases and controls
To determine if any of the genetic variants identified in case patients were associated with susceptibility or resistance to infection with WNV, we tested eight SNPs (Table 1) among all the participants enrolled in the study. The cohort of Ashkenazi Jewish participants enrolled in the study included symptomatic WNV-positive patients (n = 39) and WNV-negative controls (n = 61). There was no correlation between sex and any of genetic variants tested in the study.
A contingency table analysis based on the allele level showed that two SNPs were associated with WNV infection; rs7280422 (MX1) [odds ratio (OR) 4·05, 95% confidence interval (CI) 2·04–8·03, P < 0·0001] with the variant allele (G) found more often in cases than controls (Table 4), and rs3213545 (OASL) (OR 1·85, 95% CI 1·03–3·3, P = 0·028) (Table 4) with the variant allele (T) found more frequently in cases than controls. At the genotype level, the CG genotype of rs7280422 (MX1) was found more often in cases than controls (OR 3·6, 95% CI 1·7–7·1, P < 0·001), and similarly the TT genotype of rs3213545 (OASL) was found more often in cases than controls (OR 2·8, 95% CI 1·0–2·7, P = 0·04) (Table 4). Assuming a dominant model of inheritance only two SNPs were found to be significantly associated with WNV infection; rs7280422 (MX1) (OR 10·5, 95% CI 4·02–27·8, P < 0·0001) and rs2066786 (RFC1) (OR 2·8, 95% CI 1–8·4, P = 0·04). Assuming a recessive model only one SNP was found to be significantly associated with WNV infection, rs3213545 (OASL) (OR 4·4, 95% CI 1·3–15·5, P = 0·016).
Table 4. Contingency table analysis of eight single nucleotide polymorphisms (SNPs)

Interaction between covariates
In the multivariate logistic regression analysis we included all the covariates that were found to be significantly associated with WNV infection. The following covariates were included: SNPs: rs7280422 (MX1), rs3213545 (OASL), and rs2066786 (RFC1). A statistically significant interaction was identified only between rs7280422 (MX1) and rs3213545 (OASL). For those with the variant allele (G) at rs7280422 (MX1) and the variant allele (T) at rs3213545 (OASL), compared with the reference alleles at both sites, the odds ratio for being infected with WNV was 18·9 (95% CI 5·9–60·6).
DISCUSSION
The current study shows that some genetic variants are significantly found at higher frequencies in patients with WNV infection. Our initial hypothesis was that Ashkenazi Jews are genetically predisposed for symptomatic infection, possibly due to higher frequencies of CCR5Δ32 mutation in the chemokine receptor gene CCR5. In fact, none of the case patients was homozygous to CCR5Δ32, given our small sample size it is hard to draw robust conclusions about the lack of association between CCR5Δ32 and WNV infection. We did identify two SNPs that were significantly more frequent in WNV-infected than non-infected individuals, rs7280422 (MX1 gene) and rs3213545 (OASL gene). In addition, assuming a dominant model of inheritance, SNP rs2066786 (RFC1 gene) was also found more often in WNV-infected than non-infected individuals.
The rationale for conducting studies to investigate the genetic predisposition of humans to infection with WNV has been mainly supported by two observations, first, severe invasive neurological complications occur only in a small minority of infected individuals (<1%), second, despite the clear relationship between certain risk factors such as increasing age and immunosuppression, severe invasive neurological disease has been also reported in healthy young individuals [Reference Emig and Apple36]. The eruption of WNV disease in Israel in the past decade with minimal human disease activity in neighbouring Arab countries suggests that the Israeli population is uniquely predisposed for symptomatic infection. The current study is the first study to investigate genetic susceptibility of humans to WNV outside North America.
The three genetic variants that were identified in our cohort have already been identified as associated with susceptibility to WNV infection and symptomatic disease in North American populations; rs7280422 (MX1) [Reference Bigham21], rs3213545 (OASL) [Reference Yakub16], and rs2066786 (RFC1) [Reference Loeb22]. MX1 encodes the interferon-induced GTP-binding protein Mx1. A possible association between polymorphism in the MX1 gene and viral infections has been initially suggested by animal studies showing that Mx1 protein confers antiviral properties against influenza viruses [Reference Salomon37]. The only clue for any involvement of the MX1 gene in WNV disease has been provided by a recent study from the USA that tested SNP associations with WNV infection in 422 symptomatic WNV-positive patients and 331 asymptomatic controls, it showed that SNP rs7280422 in MX1 was associated with symptomatic WNV infection [Reference Bigham21].
The association of genetic polymorphism in the oligoadenylate synthetase (OAS) gene cluster (OAS1, OAS2, OAS3, OASL) and susceptibility to WNV was in fact the first indication that host-dependent genetic factors may play a role in WNV pathogenesis [Reference Mashimo38]. The genetic susceptibility of inbred mice to severe WNV infection was mapped to a nonsense mutation (C802 T) in exon 4 of the gene encoding the 1b isoform of the 2'–5'-oligoadenylate synthetase family [Reference Mashimo38]. The association of SNP rs3213545 (OASL gene, exon 2, C→T) with WNV infection has been reported by Yakub et al. [Reference Yakub16] who analysed 33 symptomatic WNV-infected patients and 60 healthy controls. They showed that this SNP occurred at higher frequencies in case patients than controls. Our data showed that the variant allele (T) was more common in cases than negative controls [in contrast to Yakub et al. [Reference Yakub16] who found that the reference allele (C) occurred at higher frequencies in cases than controls). However, a large study that tested SNP rs3213545 in hundreds of well-characterized patients and controls did not show any significant association with WNV infection or with disease severity [Reference Lim19].
SNP rs2066786 in RFC1 (replication factor C1) was found to be significantly associated with symptomatic WNV infection along with another SNP (rs2298771 in SCN1A) in North American patients with severe neuroinvasive disease in a recent and well-conducted study that analysed a cohort of 560 neuroinvasive cases and 950 controls (symptomatic at disease onset but did not suffer from neuroinvasive disease) [Reference Loeb22]. RFC1 which encodes the large subunit of replication factor C (RFC), a five multi-protein complex that functions as a structure-specific DNA-dependent ATPase, was shown to be essential for the simian virus 40 (SV40) in vitro DNA replication system [Reference Tsurimoto and Stillman39].
It is unclear what is the functional significance of these SNPs, particularly synonymous SNPs [rs2066786 (RFC1) and rs3213545 (OASL)], and the SNP in non-coding exon [rs7280422 (MX1)]. This issue has recently been addressed by Chen et al. [Reference Chen40] who reviewed 21429 disease–SNP associations curated from 2113 publications studying human genetic associations, they found that non-synonymous SNPs and synonymous SNPs shared a similar likelihood for human disease association and they are just as likely to be involved in disease mechanisms. A number of possible explanations have been put forward, some authors suggested that synthetic associations may explain common variant effects [Reference Dickson41]. Synthetic association describes the situation where the association of a common variant with a disease is actually due to linkage disequilibrium between the common variant and other disease-promoting rare variants that happen to segregate on the same haplotype [Reference Gibson42]. Others suggested that while most synonymous SNPs are regarded as non-functional some can impact gene regulatory sequences such as promoters, enhancers, and silencers [Reference Hunt43].
The inconsistencies in the published reports concerning genetic variants associated with susceptibility to WNV infection could be attributed, in part, to the ancestral origins of North American populations [Reference Price44, Reference Perez and Hirschman45]. In addition, the selection of the control groups could explain some of these discrepancies [Reference Loeb23]. Our cohort of case patients consisted exclusively of Ashkenazi Jews (originating from Eastern Europe) which is considered a rather genetically homogeneous population, distinct from other European populations [Reference Tian46]. Nevertheless, based on published reports [Reference Yakub16, Reference Lim19, Reference Bigham21, Reference Loeb22] and publicly available data at dbSNP-Q (https://cgsmd.isi.edu/dbsnpq/) the allele frequencies of the reported SNPs in North American populations were similar to those found in our cohort of Ashkenazi Jews.
Our study has some limitations; first, the number of case patients was rather small, it is reasonable to imply that the observed genetic variants may have arisen by chance in a genetically distinct population. Nevertheless, our findings are consistent with recent and rather large studies in North American populations [Reference Yakub16, Reference Lim19, Reference Bigham21, Reference Loeb22]. Second, we selected only nine genetic variants that have been suggested to be associated with genetic susceptibility to WNV, it is possible that other genetic variants that have been tested in larger studies could be associated with susceptibility to WNV. Third, given the fact that several WNV strains circulated in Israel since the outbreak in 2000 [Reference Lanciotti9, Reference Kopel47, Reference Bin48], it is possible that strains with variable invasive potential are responsible for the observed disease patterns.
In conclusion, we showed that few genetic variants are found more frequently in WNV-infected than non-infected individuals. These findings imply that genetic susceptibility of Ashkenazi Jews to WNV may have played a role in shaping the regional epidemiology of WNV. The biological effects of these variants need to be determined.
ACKNOWLEDGEMENTS
This work was supported by the Technion-Israel Institute of Technology (grant no. 2017300).
DECLARATION OF INTEREST
None.




