


Effective typing of verocytotoxin-producing Escherichia coli O157 (VTEC O157) is essential for outbreak investigations and epidemiological surveillance. Multi-locus variable number tandem repeat analysis (MLVA) is a highly discriminatory and reproducible molecular typing method for a variety of pathogens, including VTEC O157 [Reference Lindstedt1]. Although published MLVA methods exist for VTEC/STEC O157 [Reference Lindstedt, Vardund and Kapperud2–Reference Hyytiä-Trees5], there is a paucity of information on inter-laboratory harmonization, and as yet there is no internationally standardized nomenclature. A multi-laboratory validation of a VTEC O157 scheme targeting eight tandem repeat loci in two multiplex PCR reactions [Reference Hyytiä-Trees5, Reference Hyytia-Trees6] has been reported; however, the strategy used for data comparison was only possible if a standardized protocol was used. Recently, the results of an international consensus for the development and use of MLVA for inter-laboratory surveillance were reported proposing the sequencing of a representative set of ‘calibration’ strains to normalize raw data to actual fragment sizes, thereby enabling laboratories to share data despite using different platforms, equipment and reagents [Reference Nadon7].
The Gastrointestinal Bacterial Reference Unit (GBRU) at Colindale, Public Health England, provides reference services for VTEC O157 isolates from England and Wales, and has utilized MLVA as a front-line typing tool for epidemiology and surveillance since 2008. As the majority of European E. coli O157 confirmed cases are reported by the UK and Ireland (~ 80% in 2011) [8], the ability to easily compare MLVA results within and between these countries is imperative to facilitate national surveillance and investigation of cross-border outbreaks. Therefore, the aim of this study was to use a panel of VTEC O157, previously subtyped by GBRU, to determine the reproducibility and robustness of MLVA carried out in the Scottish E. coli O157/VTEC Reference Laboratory (SERL) and to identify and address any issues hindering the comparison of data between the laboratories.
The isolates of VTEC O157 were received and typed at GBRU in 2009 and 2010. A wide range of isolates was chosen to represent frequently occurring phage types (21/28, 8, 2) together with representatives of less common types (14, 32, 34), and reflected, as far as possible, all of the alleles for each of the eight loci in the MLVA scheme. DNA was extracted using 10% Chelex resin (Bio-Rad, UK; GBRU) or Instagene Matrix (Bio-Rad; SERL). Amplification of eight MLVA loci was carried out in two quadruplex PCR reactions [Reference Hyytia-Trees6] with some modifications. The following dye labels were used in the forward primers in the amplifications: NED in VNTR3 and VNTR17; 6-FAM in VNTR34, VNTR19, VNTR9 and VNTR36 and VIC in VNTR25 and VNTR37. PCR reactions contained Invitrogen Platinum Taq (Invitrogen, UK; GBRU) or Qiagen Multiplex PCR kit reagents (Qiagen, UK; SERL). Sizing of the amplified products was on ABI 3730 (GBRU) or ABI 3130 (SERL) Genetic Analysers with 600 LIZ (Applied Biosystems, UK) as size standard, using 50 cm (GBRU) or 36 cm (SERL) capillaries and POP-7™ polymer. Data were analysed with Peak Scanner v. 1.0 (Applied Biosystems; GBRU) or GeneMapper v. 4.0 (Applied Biosystems; SERL) software, and fragment sizes were imported and analysed in BioNumerics v. 6.6 (Applied Maths, Belgium). EDL933 was included in each run as a positive control.
Loci were named and reported as proposed recently [Reference Nadon7]. Alleles were assigned based on their actual sequenced copy number, which was determined by sequencing a subset of the 67 isolates (n = 17). For sequencing, the MLVA primers were used in singleplex PCR but the forward primers were unlabelled. The PCR products were cleaned with Illustra ExoProStar 1-Step™ (GE Healthcare Life Sciences, UK) and both forward and reverse strands were sequenced using the ABI Cycle-sequencing kit v. 3.1 (Applied Biosystems). The products were cleaned and run on the ABI 3130xl automated sequencer. Sequences were analysed in BioNumerics and Tandem Repeat Finder [Reference Benson9] was used to confirm copy numbers.
Identical MLVA profiles were obtained for 65/67 (97%) isolates in both laboratories (data not shown). The two discrepancies in MLVA profile were due to single locus difference between strains. In one case, no peak was detected at ECS5426. When this locus was re-amplified using singleplex PCR and agarose gel electrophoresis, no PCR product was detected, suggesting either a failure of primer binding due to a primer binding site mutation or the loss of the locus in the strain. In the other case, a one repeat difference of 7 bp was detected at pO15754 (216 bp vs. 223 bp). The most likely explanation for these differences was the strains evolved during subculture.
As shown in Table 1, the inter-laboratory fragment sizing was very reproducible (0–2 bp) despite differences in the Genetic Analyser models, capillary column sizes, PCR reagents and run conditions. Others have previously reported inter-laboratory fragment sizing within 1·5 bp when the same platform was used, while differences of up to 6 bp have been observed when different machines were used [Reference Hyytia-Trees6].
Table 1. Details of the VTEC O157 VNTR loci*
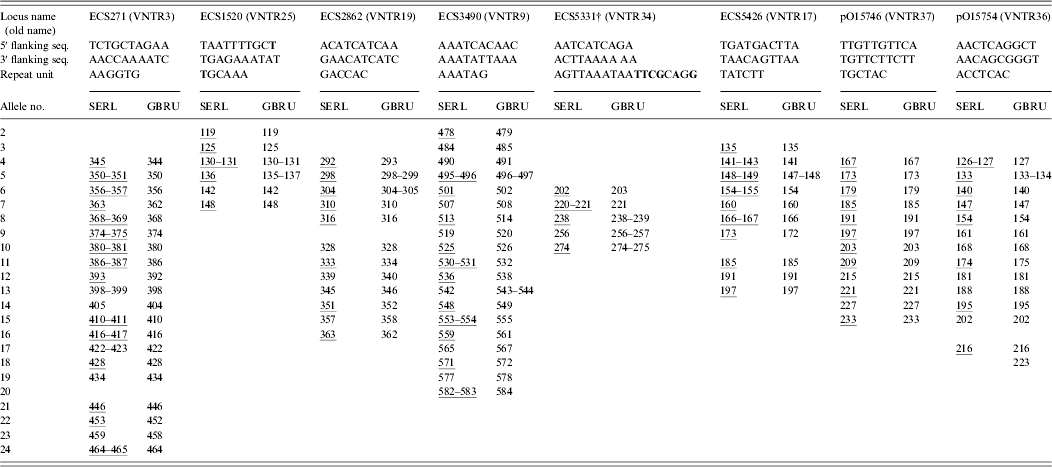
SERL, Scottish E. coli O157/VTEC Reference Laboratory; GBRU, Gastrointestinal Bacteria Reference Unit.
* Consensus 5′ and 3′ flanking sequences and repeat unit sequences are shown; sequence variation is highlighted in bold; and representative alleles that were sequenced are underlined.
† Five different repeat unit sequences were identified for ECS5331: AGTTAAATAATCTACAGA, AGTTAAATAATATACAGA, AGTTAAATAATATACAGG, AGTTAAATAATTCGCAGG, and AGTTAAATAACTCGCGGG.
The sizing discrepancies were most notable between the laboratories in locus ECS3490, probably because the difference in the capillary array lengths causes resolution differences for large fragments. Moreover, the likely reason for the overall good reproducibility between the laboratories was that the same polymer type was used for fragment analysis. Differences in the composition of polymers cause different levels of secondary and tertiary structure in the fragments which in turn cause the fragments to migrate differently through the polymer resulting in notable fragment size discrepancies [Reference Rosenblum10].
To determine allele nomenclature we sequenced a subset of 17 genetically diverse strains covering a representative set of alleles for each of the eight loci, which are highlighted in Table 1. As proposed by Nadon et al. [Reference Nadon7], the smallest, largest and at least every third allele of each locus were sequenced. We found the repeat units were homogeneous in size; however, incomplete repeats were observed. Most notably, ECS3490 loci always ended with an incomplete repeat of 0·8. According to the proposed criteria, incomplete repeats should be rounded down to the nearest complete copy number, therefore the ECS3490 allele for EDL933 was assigned 10, rather than 11 as previously reported [Reference Hyytiä-Trees5, Reference Lindstedt11]. The full MLVA profile for EDL933 was 9-5-6-10-10-6-7-8. To help identify the start and end of the repeat regions, the 5′ and 3′ sequences immediately flanking the tandem repeats were identified (see Table 1). Sequence variation was observed in the 5′ flanking sequences of ECS1520 in two of the 17 strains tested (TAATTTTGCT→TAATTTTGCC). Sequence variation was also identified in one of the ECS1520 repeat units in one strain (TGCAAA→AGCAAA), and in all sequenced strains five different repeat unit sequences were identified for ECS5331 (see Table 1).
Problems identified in comparing MLVA data produced in different laboratories have been reported previously [Reference Hyytia-Trees6]. In this study, we used one of the MLVA protocols available for VTEC O157 and followed recent guidelines based on international consensus to name alleles based on actual copy numbers enabling the successful comparison of data generated in our laboratories, despite the use of different equipment and reagents. Similar work has been reported for Salmonella Typhimurium MLVA [Reference Larsson12, Reference Larsson13]. Since the completion of the present study, we have exchanged real-time MLVA data to aid cross-border outbreak investigations, and are implementing a quality assurance programme to ensure the continual provision of high-quality typing data, which could be extended to other countries. The panel of 17 reference strains can be provided to other laboratories interested in calibrating their VNTR data to actual sequenced copy numbers and is available for inclusion in an extended European or international calibration set. The international harmonization of MLVA for VTEC would facilitate the development of a central database and a consensus nomenclature for allelic profiles to enable the rapid identification of known or novel strains for international surveillance and outbreak detection of VTEC.
ACKNOWLEDGEMENTS
The authors thank Leila Shaw and Tracy Dransfield for technical assistance.
DECLARATION OF INTEREST
None.

