INTRODUCTION
Amoebiasis, caused by the intestinal parasite Entamoeba histolytica, has an estimated worldwide prevalence of 500 million cases of symptomatic disease, and 40 000–110 000 deaths annually. Amoebiasis is the third leading parasitic cause of death worldwide [Reference John, Salata, Kliegman, Stanton, St. Geme, Schor and Behrman1, Reference Pham2]. It is an important health problem, especially in developing countries [Reference Haque3]. The rate of infection by E. histolytica differs among countries, socioeconomic status, sanitary conditions and populations [Reference Al-Harthi and Jamjoom4]. It is highly endemic throughout the poor and socioeconomically deprived communities in the tropics and subtropics. Environmental, socioeconomic, demographic and hygiene-related behaviours are known to influence the transmission and the distribution of intestinal parasitic infections [Reference Norhayati5].
Reliable clinical diagnosis of invasive amoebiasis has important epidemiological implications, since the majority of official epidemiological sources of statistics on health, particularly in developing countries, is based on clinical observations [Reference Ximenez6]. Major problems with a wide array of imperfect tests for the diagnosis of amoebiasis severely limit the understanding of its magnitude and epidemiology. A greater hindrance is the varied, inconsistent application of existing methods in different areas of the world.
The sensitivity of light microscopy is limited to only 60% at best, but is confounded by false-positive results due to mis-identification of macrophages and non-pathogenic species of Entamoeba [Reference Haque7]. Amoebic culture with isoenzyme analysis is considered to be a reference standard to differentiate E. histolytica from E. dispar, but this method is not widely available and is not practical for routine diagnostic laboratories [Reference Haque7]. Moreover, factors such as delay in processing stool specimens and initiation of anti-amoebic treatment prior to sample collection can lead to E. histolytica-negative culture results even in stool samples showing the presence of cysts/trophozoites by light microscopy [Reference Khairnar, Parija and Palaniappan8]. A study conducted on children by Petri and co-workers in Bangladesh revealed the limitations in microscopic studies, where only 40% of microscopically positive cases were proven to have E. histolytica infection when tested by polymerase chain reaction (PCR)-based methods [Reference Petri9]. Currently this technique is widely used to detect Entamoeba species because of their high sensitivity, being 100 times more sensitive than ELISA [Reference Fotedar10, Reference Mirelman, Nuchamowitz and Stolarsky11].
The progressive decline and ultimate collapse of immune system functions, which are characteristic of AIDS, usually results in morbidity and ultimately death due to opportunistic bacterial, viral, and parasitic infections [Reference Durack12]. Parasitic infection is still a major problem in patients suffering from HIV+/AIDS throughout the world [Reference Savioli13]. The incidence of parasitic infections was 50% in developed countries while it reached 95% in developing countries [Reference Nkenfou, Nana and Payne14]. The interpretation of these studies, however, is difficult because the majority of the diagnoses of amoebiasis was based solely on microscopic examination of stool samples, which is an insensitive test that fails to adequately discriminate between E. histolytica and E. dispar [Reference Tanyuksel and Petri15]. In the present study, we have systematically developed a protocol to detect and differentiate Entamoeba species (E. histolytica and E. dispar) in a population. We further compared the sensitivity of the techniques using molecular probes. We have also evaluated and compared the load of the two species in HIV-positive and HIV-negative groups and the risk factors, if any.
METHODS
Patient samples and demographic parameters
The study involved a total of 643 stool samples. The samples were classified into two groups that included 178 HIV-positive samples and 465 HIV-negative samples. In the HIV-positive group, 94 non-diarrhoeal patients volunteered to submit samples and in the HIV-negative group, 70 people who were without any diarrhoeal complaints volunteered to submit samples. The latter group mostly included the attendants of the patients, their family members and the hospital staff who did not complain of any diarrhoeal episodes. Two different locations were studied. (1) Patients with diarrhoeal complaints submitted their samples to the outpatient department (OPD) of Pathology at Safdarjung Hospital, New Delhi under the supervision of the pathologist. (2) From Assam there were two groups: (a) non-HIV samples were collected from two hospitals, Silchar Medical College and Hospital (SMCH) and Nari Shikha Hospital, Silchar, Assam; (b) HIV samples were collected from the Antiretroviral Therapy (ART) Centre of SMCH, Silchar, Assam. Samples of HIV-positive patients were collected for two groups, i.e. with and without ART.
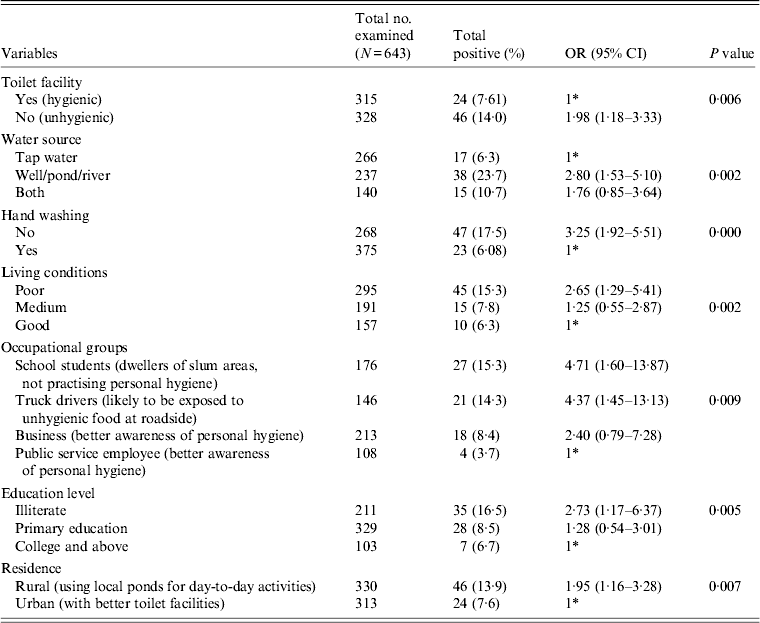
All the stool specimens were subjected to pathological examination for the presence of any enteric parasites, e.g. Giardia, Blastocystis hominis, Entamoeba coli, E. hartamanii besides E. histolytica or E. dispar. The ethical approval for sample collection was obtained from the Institutional Ethical Committee (IEC), Gurucharan College, Silchar, Assam and the Institute Ethical Research Board (IERB) of Jawaharlal Nehru University, New Delhi before the commencement of the study. Out of the above samples, 178 patients were recruited in the HIV-positive group and 465 patients were recruited in the HIV-negative group. The samples were collected following a standard questionnaire prepared for the study that included demographic features (Table 1) and various risk factors that may contribute to the prevalence of the disease (Table 2). The attending physician and the research personnel completed the questionnaires in this study during sample collection. The trained research assistants interviewed participants in person, asking questions on demographic data (e.g. age, gender, education level), socioeconomic background (e.g. occupation, household income, educational status), behavioural risks (e.g. personal hygiene such as hand washing and food consumption), environmental sanitation and living condition characteristics (e.g. types of water supply, latrine system, sewage disposal system, etc.). The subjects were further classified on the basis of HIV status to see if any change in the prevalence rate was observed (Table 1).
Table 1. Prevalence and association of E. histolytica and E. dispar according to HIV status and other demographic features

OR, Odds ratio; CI, Confidence interval; ART, antiretroviral therapy.
* 1 = Reference category.
Table 2. Association between E. histolytica and E. dispar and the various risk factors
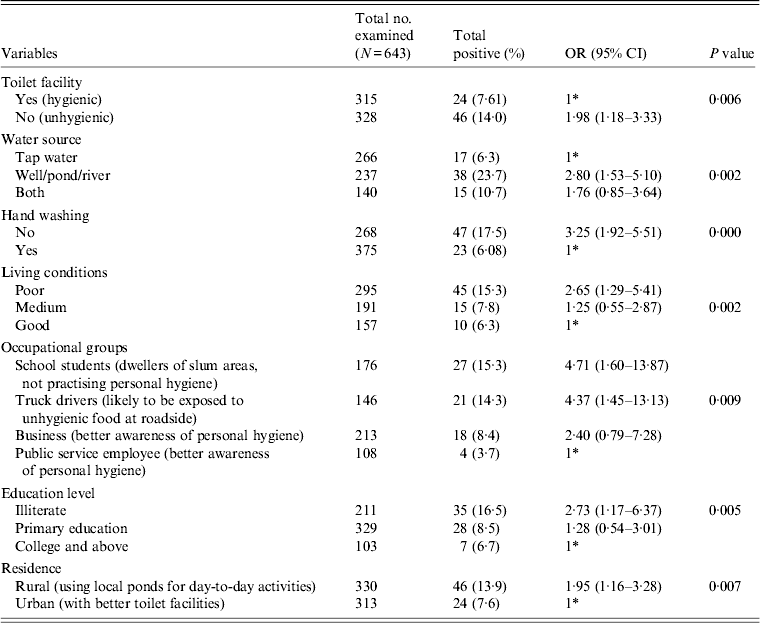
OR, Odds ratio; CI, Confidence interval.
* 1 = Reference category
Sample size determination
The expected sample size was calculated according to the following parameters: expected prevalence of E. histolytica/E. dispar varied between 8% and 15% at a confidence interval of 95% and absolute precision (d) = 0·05 [Reference Daniel16]. The minimum sample size needed in this study was around 113–200. The research team decided to increase the total sample size by three times to increase the power of the study in order to guard against incomplete data. We recruited 178 individuals in the HIV-positive group and 465 in the HIV-negative group for the study. We conducted a cross-sectional study during the period April 2011 to June 2013.
 $$_\; n = \displaystyle{{Z^2 P(1-P)} \over {d^2}}, $$
$$_\; n = \displaystyle{{Z^2 P(1-P)} \over {d^2}}, $$
where n = sample size, Z = Z statistic for a level of confidence, P = expected prevalence or proportion (in proportion of 1; if 15%, P = 0·15), and d = precision (in proportion of 1; if 5%, d = 0·05). Z statistic (Z): for the conventional level of 95% confidence, Z value is 1·96.
HIV-positive confirmation
Subjects were confirmed as HIV positive at the Integrated Counselling and Testing Centre, SMCH, Silchar by three successive serological rapid tests using the Combs Aids, Pareekshak® HIV ½ Triline card test (Bhat Bio-Tech, India) and SD Bioline anti-HIV ½ test kits (Standard Diagnostics Inc., India), according to the manfacturer's instructions. The CD4+ T cell count was performed using CyFlow® Counter (Partec, Germany).
Stool sample collection and processing
Fresh stool samples were collected from the study participants (n = 643). The samples were collected in wide-mouth screw-capped containers, pre-labelled with the individual's name and code and were distributed to each participant for the collection of around 5 g faecal sample. The samples were collected within 2 h of defecation and delivered to the laboratory. The samples were processed following the protocol described in Figure 1.

Fig. 1. Systematic approach for determination of the prevalence rate of Entamoeba spp. (E. histolytica and E. dispar).
Screening of samples by dot-blot
Initial screening of the samples was done by the dot-blot technique which identifies both E. histolytica and E. dispar using a probe specific to both.
Enrichment of Entamoeba cysts
The cysts were enriched following the protocol of Knight et al. [Reference Knight17], with slight modifications. Briefly, faecal samples (1–2 g) were homogenized in 10 ml autoclaved distilled water, and strained through cheesecloth in a 50 ml Falcon tube. This suspension was centrifuged at 1200 g for 3 min and the pellet was re-dissolved in 10 ml of 10% formaldehyde and incubated for 15 min at room temperature. Next, 3 ml of diethyl ether was added to the tube and this mixture was vortexed and incubated at room temperature for 30 min. The mixture was subjected to centrifugation at 1200 g for 3 min, the supernatant was removed and pellet was washed with double-distilled water. The pellet containing concentrated cysts was re-dissolved in 400 μl T10E1 buffer. The cysts in T10E1 buffer were subjected to freeze–thaw cycles and thereafter to sonication in order to obtain crude DNA for the dot-blot hybridization experiments.
Dot-blot hybridization
The crude cyst DNA was denatured by the addition of NaOH to a final concentration of 0·25 n in a total volume of 300 μl. The DNA was kept at room temperature for 30 min and then transferred onto ice. The GS+ nylon membrane of required size was cut and saturated in 0·4 m Tris-Cl (pH 7·5) for 15 min. Thirty samples could be loaded in triplicate at one time along with appropriate controls. Twenty nanograms of DNA was spotted onto the membrane with the help of a mini-fold apparatus (96-well platform) from Whatman, Germany. The blots were air dried and UV cross-linked before hybridization. A 4·5 kb rDNA fragment (EcoRI to HindIII site) from the HMe region of EhR1 (rDNA plasmid in HM1:IMSS strain of E. histolytica) was used as a probe [Reference Srivastava, Bhattacharya and Paul18] for detection of Entamoeba-positive samples that include both E. histolytica and E. dispar (Fig. 2 a). The probe hybridizes with both E. histolytica and E. dispar, but not with other Entamoebae (E. moshkovskii and E. invadens) [Reference Srivastava, Bhattacharya and Paul18].

Fig. 2. Diagram of screening of stool samples by dot-blot methods. (a) Linear map of EhR1 episome (24·5 kb) showing the position of the HMe probe (4·5 kb) common for both E. histolytica and E. dispar. (b) Diagram of dot-blot analysis of stool sample using the HMe probe. Rows 1–5 (columns A–D) represent spots of DNA from stool samples. Rows 7 and 8 are blank. Row 6, column A: E. histolytica HM1:IMSS genomic DNA as positive control; column B: E. dispar genomic DNA as positive control; column C: plasmid with cloned HMe as positive control; column D: negative stool DNA as negative control.
Genomic DNA extraction from stool sample
The genomic DNA was purified from the frozen stool samples using the QIAamp DNA stool kit (Qiagen, USA) according to the protocol provided by the manufacturer, with some modifications. Briefly, following five freeze–thaw cycles, samples were incubated at 95°C for 10 min in ASL buffer (Lysis buffer, Qiagen). The samples were then processed according to the manufacturer's instructions.
PCR assay
Dot-blot-positive samples were subjected to further validation by PCR. This assay was based on the amplification of the Intergenic spacer region using primers from the end of 18S and the start of the 28S rRNA gene of E. histolytica/E. dispar (Fig. 3). The primary PCR for the detection of Entamoeba genus was performed using a primer set described in Table 3, in a 25 μl reaction containing 2·5 μl of 10 × PCR buffer, 0·5 μl of 0·2 mm dNTPs, 1·5 μl of 2·5 mm MgCl2, 2·5 μl of 10 × BSA, 0·5 μl of 20 pmol of each primer, 0·25 μl of 2·5 U Taq polymerase and 200 ng DNA template. Nuclease free water was added to a final volume of 25 μl. The reaction was performed with an initial denaturing step at 94°C for 5 min followed by 30 cycles of 94°C for 30 s (denaturation), 51°C for 30 s (annealing), 72°C (extension) for 45 s and a final extension for 7 min at 72°C.

Fig. 3. Diagram showing identification of Entamoeba spp. (E. histolytica and E. dispar) by PCR methods. (a) Schematic representation of location of genus-specific primers. (b) Detection of both species of Entamoeba (E. histolytica and E. dispar) using genus-specific primers. Lane 1, E. histolytica genomic DNA used as positive control; lane 2, E. dispar genomic DNA used as positive control; lanes 3–6, stool DNA samples positive for E. histolytica or E. dispar; lane 7, E. coli genomic DNA used as negative control.
Table 3. List of primers and probes used in the study

Subsequently, Entamoeba species-specific PCR was performed by amplifying the following primer sets as described in Table 3. E. histolytica was amplified using SINE2 primers and E. dispar was amplified using the forward primer from the 18S region and the reverse primer from the ITS2 region of the rDNA molecule [Reference Srivastava, Bhattacharya and Paul18] (Fig. 4). Cycling conditions for the secondary amplification were the same as for the primary reaction except the annealing temperature, which was 55 °C for E. histolytica and 40°C for E. dispar. Extension time for E. histolytica and E. dispar were 30 s and 1 min 20 s, respectively. All the control DNA samples were subjected to the same conditions. The PCR products were analysed on 1·0% agarose gel.

Fig. 4. Diagram showing identification of species by PCR method. (a) Schematic representation of E. histolytica-specific primers located in the SINE2 (EhSINE2) element of E. histolytica. (b) Detection of E. histolytica in stool samples using E. histolytica-specific primers. Lane M, 100 bp marker; lane 1, E. histolytica genomic DNA as positive control; lanes 3, 5, 6, 8, stool genomic DNA positive for E. histolytica; lanes 2, 4, 7, stool genomic DNA negative for E. histolytica; lane 9, no template control. (c) Schematic representation of E. dispar-specific primers located on rDNA molecule. (d) Detection of E. dispar in stool samples using E. dispar-specific primers. Lane M, 1 kb marker; lane 1, E. dispar genomic DNA used as positive control; lanes 2, 3, 6, stool genomic DNA positive for E. dispar; lanes 4, 5, 7, stool genomic DNA negative for E. dispar; lane 8, no template control.
Data analysis
Statistical analysis was performed using SPSS v. 16.0 software (SPSS Inc., USA). Mean, range and percentage were used to describe the different characteristics of the study subjects as appropriate. The qualitative data were presented in the form of number and percentage. Pearson's χ 2 test was used to test the associations between each variable in a univariate statistical model, with prevalence of E. histolytica/E. dispar as a dependent variable, while the independent variables were HIV status, CD4 cell count, gender, age group and other sociodemographic profiles. The odds ratio (OR) and 95% confidence interval (CI) were computed by univariate logistic regression analysis for statistically significant factors showing a level of statistical significance of P < 0·05.
RESULTS
The screening technique employed here in Figure 2 illustrates one of the dot-blot tests, showing the positive spots for both the species that hybridized with the HMe probe. Genomic DNA isolated from the axenic cultures of HM1 (E. histolytica) and E. dispar served as positive controls and the stool sample DNA negative for E. histolytica or E. dispar was considered as a negative control. Crude DNA extracted from the stool samples were loaded on the dot-blot manifold apparatus. Twenty-five samples in triplicate were spotted onto each blot. Our data show that out of a total of 643 samples analysed, 75 (12%) samples hybridized with the probe. As expected, the distribution of positive spots was higher in the HIV-positive group (15%) compared to the negative group (9%).
The positive samples from the dot-blot assays were further subjected to validation by designing genus-specific primers from the ITS region of the rDNA molecule of Entamoeba. The location of the primers and the sizes of the amplicons are shown in (Fig. 3). The primers designed here, amplified a 609 bp region when pure genomic DNA of either E. histolytica or E dispar was used as template as shown in lanes 1 and 2 (Fig. 3). Out of 75 dot-blot-positive samples, 70 exhibited a positive amplification with these sets of primers. Five samples were again repeated for amplification, but did not yield a positive result. In order to test the sensitivity of the technique, we also validated our results by PCR with a few DNA samples that were dot-blot negative. Following the method of Giles et al. [Reference Giles19], the sensitivity and specificity of the dot-blot analysis was found to be 100% and 99%, respectively, whereas by the PCR method using genus-specific primers, it was 100% for both sensitivity and specificity [Reference Giles19].
Further confirmation at the species level was carried out using E. histolytica-specific and E. dispar-specific primers (see Fig. 4). All the 70 samples that yielded a positive amplification using the genus-specific primer were subjected to further identification using SINE2 primers for E. histolytica and 18S/ITS2 primers exclusive for amplifying E. dispar [Reference Srivastava, Bhattacharya and Paul18]. The amplicon sizes were 350 bp and 1·3 kb, respectively. The PCR assay was also tested for specificity against a panel of DNAs extracted from 12 faecal samples, each containing one or a mixture of the following parasites as revealed by microscopy: Blastocystis hominis, Escherichia coli, Giardia intestinalis, Endolimax nana, Iodamoeba butschlii, Cryptosporidium sp. Our results demonstrated 39/643 as positive for E. histolytica when all the samples were combined. The distribution of E. histolytica in the HIV-positive and HIV-negative groups was 11/178 and 28/465, respectively, whereas for E. dispar, it was 9/178 and 11/465, respectively (Table 4). We also observed mixed infections in a few cases, seven in HIV-positive and four in HIV-negative individuals. In the HIV-positive samples, most (n = 102) were on antiretroviral therapy (ART) and only 76 were not receiving ART. The PCR analysis of our samples recorded the highest rate of infection (23·68%) in the group of patients without ART compared to the group receiving ART (8·82%).
Table 4. Summary of mono and mixed infections of E. histolytica and E. dispar detected by PCR assay

PCR, Polymerase chain reaction; ART, antiretroviral therapy.
Various demographic features that contributed to the prevalence of the parasite are compared in Table 1. Our statistical analysis revealed that the association of the parasite was significantly higher in the HIV-positive (P = 0·031) compared to the HIV-negative group. The HIV-positive group showed a higher value (19%) of parasite load in the symptomatic group compared to the asymptomatic group (11%); however, this did not attain a significant P value. A similar pattern was also observed in the non-HIV group. The prevalence rate of the parasite was found to be significantly higher in the HIV-positive group (P = 0·011) without ART compared to patients receiving ART. When we compared the role of age on the prevalence of the parasite, we did not find a significant association for the HIV-positive group, whereas for HIV-negative group a significant association was observed (P = 0·015) in the <15 years age group. Further, we did not observe any significant association of gender in either the HIV-positive or HIV-negative groups with parasite load. As expected, a significant increase in the prevalence of the parasite (P = 0·008) was observed in the HIV-positive group with a CD4 cell count <200. We observed a significant impact of season on the prevalence of the parasite. During the monsoon season (June–September), the parasite load was significantly higher (~15%) (OR 2·42, 95% CI 1·29–4·54, P = 0·017) compared to the pre- or post-monsoon seasons.
Table 2 gives the results of the logistic regression analysis showing the association between the parasite and the various risk factors contributing to the infection. Our data revealed that unhygienic toilet facilities contributed to the positive cases with a significant P value of 0·006 (OR 1·98). When the water sources used for day-today activities were analysed, people using river water or pond water were found to be more prone to the infection (OR 2·80, 95% CI 1·53–5·10, P = 0·002) compared to the group using tap water for the same purpose. Our data further show that the individuals with the habit of washing hands before eating are less prone to infection compared to the group lacking this habit. We observed a significant association of the parasite with the latter group in our study (OR 3·25, 95% CI 1·92–5·51). Poor quality of living conditions significantly contributed to the infection rate and attained a P value of 0·002 compared to medium and good quality of living conditions of the inhabitants under study. Among other risk factors, as in various occupational groups, the school students (OR 4·7, P = 0·009), followed by truck drivers (OR 4·3) and the business group (OR 2·4) were at higher risk compared to the public service employee group. Our analysis further showed a significant association of the disease with the education level of respondents with a P value of 0·005 in the illiterate group (OR 2·73), and the group with primary education (OR 1·28) compared to the college-educated group. In our study group, almost 50% of respondents represented the rural population and were significantly associated with the disease (OR 1·95, P = 0·007) compared to the urban population.
DISCUSSION
E. histolytica is morphologically identical to E. dispar and the two species can only be distinguished by molecular, biochemical and antigenic differences [Reference Diamond and Clark20, Reference Haque and Petri21]. Therefore, microscopy technique to estimate the parasitic load accurately has always remained controversial.
Through the above systematic approach of diagnosis of Entamoeba, we have been able to develop a cost-effective and high-throughput sensitive technique for screening large numbers of stool samples at one time. Dot-blot hybridization can screen Entamoeba-positive stool samples from a large pool of samples. The HMe probe (4·5 kb), designed in our laboratory specifically hybridized the rDNA fragment generated from the intergenic spacer region of the rDNA molecule of E. histolytica or E. dispar [Reference Paul, Bhattacharya and Bhattacharya22]. In the present technique a crude extract of genomic DNA was isolated from the formalin-ether concentrated cysts from stool samples. Our analysis revealed five false-positive samples that did not amplify when dot-blot-positive samples were further subjected to PCR using genus-specific primers. The dot-blot technique was found to have 100% sensitivity and 99% specificity. However, it saves the cost of two important steps which is very relevant in developing diagnostics for parasites in endemic countries like India. It minimizes (1) the cost of a large number of stool kits for isolating genomic DNA from the stool samples and (2) helps to scale down the number of subsequent PCR reactions needed to identify the parasite at the genus or species level. The genus-specific primers were designed in the present study to amplify the ITS1 and ITS2 regions between the 18S and 28S loci of the rDNA molecule. These primer sets successfully amplified the dot-blot-positive samples with high sensitivity and specificity and did not amplify any dot-blot-negative sample (data not shown) or any bacterial DNA present in the stool sample as shown in Figure 3. The sensitivity of the PCR technique applied to distinguish the two species of Entamoeba has been described previously [Reference Srivastava, Bhattacharya and Paul18]. In our analysis, out of a total of 643 samples we performed PCR reactions for only 70 samples; this was possible as we had introduced a screening technique at the outset. We performed a cross-sectional study of our population in two different locations of India; however, our data did not show any significant differences in the prevalence rate (data not shown) of the parasite. The overall prevalence rate of the parasite observed in our study using the molecular technique was 10·89%. A prevalence rate of 18·6% of Entamoeba was observed in a Malayasian population using a microscopic technique [Reference Anuar23]. Using a molecular technique, Ngui et al. reported a prevalence rate of 17·6% [Reference Ngui24]. A study conducted in an Australian population revealed the significance of molecular characterization of Entamoeba over microscopy methods [Reference Fotedar10]. However, we observed a significant fluctuation in the prevalence of the parasite on the basis of the HIV status of the patients. As expected, with the decrease in the CD4 cell count in the HIV-positive patients, a significant increase in parasite load was observed, supporting the earlier observations made in Ethiopian and Mexican populations [Reference Adamu, Wegayehu and Petros25, Reference Morán26]. Our study further confirms that ART contributed significantly in protecting individuals from parasitic infection. Since Entamoeba may exist in both symptomatic and asymptomatic states in the intestine, we therefore concentrated on a few asymptomatic individuals in both the HIV-positive and HIV-negative groups using our molecular probes and the primers specific for the parasite. Our result also suggested no significant change in the prevalence of the parasite in either group. This requires further confirmation on a larger dataset. Samples collected both from Northern India and Eastern India revealed that the prevalence of the parasite was highest in the monsoon season followed by the pre- and post-monsoon seasons. The χ 2 test revealed a significant P value of 0·017. This further indicated the high rate of faecal–oral contamination during the monsoon season, probably due to lack of safe drinking water. The prevalence was significantly high in the <15 or >45 years age groups for the non-HIV group. However, such a correlation was not observed in the HIV-positive group, perhaps due to unavailability of sufficient samples in the different age groups. The trend emerging from our analysis indicates that the various risk factors, e.g. toilet facilities, living conditions, hygienic practices, rural background of respondents, sources of drinking water, occupation and education level are important predictors of parasite prevalence. Such a correlation was also drawn from a study conducted in an agricultural community of Vietnam and in rural residents of Kenya [Reference Pham2, Reference Kinuthia27].
The systematic approach attempted in the present study reveals that the dot-blot assay can offer an economic and faster way of screening Entamoeba-positive samples from a large number of stool samples and, therefore, could be a promising tool besides the PCR technique that requires a more elaborate infrastructure. Our study also concluded that the PCR-based technique should be made available to clinics to delineate the two species to avoid overuse of the drug metronidazole. From this study we further concluded that ART can reduce the prevalence of the parasite considerably in the HIV-positive group. Therefore, ART should be made available to HIV-positive patients at an early stage to avoid parasitic infections. Risk factors like poor sanitation, contaminated drinking water, poor hygienic habits and rural background contributed to the disease and these need to be curbed in order to reduce the prevalence of the parasite.
ACKNOWLEDGEMENTS
The authors acknowledge the subjects who participated in the study. We also thank the hospital staff who helped with microscopic observations and sample collection. The work was supported by a research grant to J.P. and B.S. from the Department of Biotechnology, Government of India (BT/55/NE/TBP/2010) and partially by a Molecular Parasitology project to J.P. from the Department of Biotechnology, India (COE- BT/01/CEIB/11 V/08).
DECLARATION OF INTEREST
None.