


Inflammation in health and disease
Inflammation is an essential and normal component of the immune response that protects against pathogenic organisms and is involved in the response to injury(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) . In general, inflammation acts to create an environment that is hostile to pathogens, it initiates pathogen killing, and it causes changes in the metabolism of the host(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) . Many cell types play roles in the inflammatory response, which involves the production of, and responses to, a vast number of chemical mediators(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) . The cardinal signs of inflammation are redness, swelling, heat, pain and loss of function. These are caused by the cellular activation and chemical mediator release that occur during the initiation and perpetuation of the inflammatory response. The chemical mediators released from cells during inflammation include lipids (e.g. PG, leukotrienes (LT), endocannabinoids, platelet-activating factor), peptides (e.g. cytokines, chemokines), reactive oxygen species (e.g. superoxide anion, hydrogen peroxide), amino acid derivatives (e.g. histamine, nitric oxide) and enzymes (e.g. matrix proteases) depending upon the cell types present, the nature of the inflammatory stimulus, the anatomical site involved and the stage during the inflammatory response(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) . Although the inflammatory response is designed to be damaging to pathogens, the cellular activities involved and the chemical mediators produced can cause damage to host tissues(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) . Fortunately therefore, inflammation is normally self-limiting and resolves, often rapidly. This is because various inhibitory mechanisms are activated as inflammation runs its course(Reference Barnig, Bezema and Calder3). These include shedding of receptors for pro-inflammatory cytokines and increased generation of anti-inflammatory cytokines(Reference Barnig, Bezema and Calder3). Another mechanism involved is the generation of specialised pro-resolving lipid mediators (SPM) which act to inhibit pro-inflammatory signalling(Reference Serhan4). Loss of the regulatory processes involved in the resolution of inflammation can result in excessive, inappropriate or on-going inflammation that can cause irreparable damage to host tissues leading to pathology and disease (Fig. 1)(Reference Barnig, Bezema and Calder3–Reference Innes and Calder5). Inflammation is an important component of a wide array of human conditions including classic chronic inflammatory diseases such as rheumatoid arthritis (RA), inflammatory bowel diseases, multiple sclerosis, lupus, chronic obstructive pulmonary disease, allergy and asthma which are all controlled or treated with varying degrees of success with anti-inflammatory medications(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) . Research in the past two decades has identified that inflammation is also a risk factor involved in atherosclerosis(Reference Glass and Witztum6,Reference Hansson7) , cardiovascular events(Reference Glass and Witztum6,Reference Hansson7) , neurodegenerative disorders and cognitive decline(Reference Novellino, Saccà and Donato8), and in many cancers(Reference Colotta, Allavena and Sica9). Furthermore, an excessive inflammatory response is linked to adverse outcomes following surgery(Reference Shankar Hari and Summers10) and in critical illness(Reference Ramírez, Ferrer and Martí11). Inflammation appears to be a driver of loss of lean mass seen in many cancers(Reference Fonseca, Farkas and Dora12) and in frailty(Reference Vatic, von Haehling and Ebner13) and sarcopenia(Reference Livshits and Kalinkovich14). Finally, both obesity and ageing are associated with an elevated state of inflammation(Reference Calder, Ahluwalia and Brouns15,Reference Calder, Bosco and Bourdet-Sicard16) and this might relate to the development of chronic conditions associated with both. Because of this wide-reaching adverse impact of inappropriate or excessive inflammation, strategies that control or resolve the inflammatory response could have a huge impact on human health and well-being. This review will describe mechanisms by which fatty acids can influence inflammatory processes with a focus on the effects of n-3 PUFA EPA and DHA. An overview of the structure, metabolism, dietary sources and intakes, and general health benefits of EPA and DHA is available elsewhere(Reference Calder17).
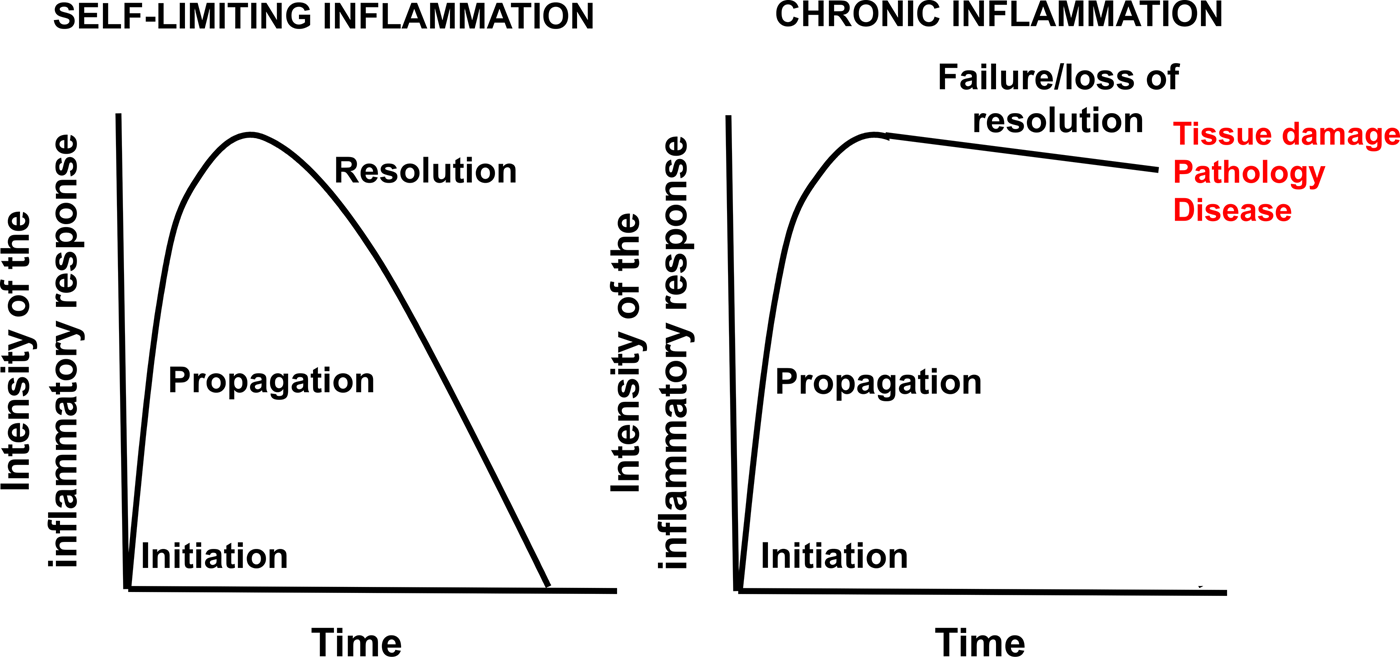
Fig. 1. (Colour online) Schematic representation of self-limiting and chronic inflammation. Modified from Prostaglandins Leukotrienes and Essential Fatty Acids, Vol 131, J.K. Innes and P.C. Calder, Omega-6 fatty acids and inflammation, pp. 41–48, Copyright 2018, with permission from Elsevier(Reference Innes and Calder5).
The links between fatty acids, cell membranes and inflammation
Many of the receptors for triggers of inflammation (e.g. several Toll-like receptors (TLR) that recognise microbial structures or products such as lipopolysaccharide (LPS)) and for inflammatory mediators such as cytokines, chemokines, PG and LT are localised in the cell membrane. Here the receptors interact with other membrane proteins to initiate intracellular signalling leading to cellular activation. In many cases, proteins need to move within the plane of cell membrane to come together and form signalling platforms. Such movement within the membrane will obviously involve some form of interaction with the lipid components of the membrane. The term lipid rafts has been used to describe the signalling platforms that result, and these structures include the various proteins and their surrounding lipids, which are often specific species of phospholipids, sphingolipids and cholesterol(Reference Sviridov, Mukhamedova and Miller18). Once rafts form, they generate signals that regulate how cells respond. For example, in monocytes and macrophages, binding of LPS to TLR4 first initiates the formation of rafts involving other proteins including myeloid differentiation primary response gene 88 (MyD88)(Reference Ruysschaert and Lonez19); the proteins within this raft structure in turn generate the signals that ultimately activate NFκB. Activation of NFκB causes its translocation to the nucleus where it binds to the response elements in a number of genes, including those encoding classic inflammatory cytokines such as TNF, IL-β, chemokines such as IL-8, the PG-producing enzyme cyclooxygenase (COX) 2, inducible nitric oxide synthase, adhesion molecules and matrix metalloproteinases(Reference Kumar, Takada and Boriek20). Ultimately these proteins are produced, establishing and perpetuating the inflammatory response. NFκB is also activated by other inflammatory stimuli including inflammatory cytokines, oxidative stress and UV irradiation. It is important to note that NFκB is just one of a number of transcription factors and signalling pathways involved in the inflammatory response.
It seems likely that the fatty acid composition of inflammatory cell membrane phospholipids will influence the ability of proteins to move within the plane of the cell membrane and will affect the ability of lipid rafts to form (this will be discussed later with regard to DHA). What this means is that modifying the fatty acid composition of the membranes of cells involved in inflammation could influence the earliest events in inflammatory signalling. There are likely to be at least two other impacts of altering cell membrane fatty acid composition in these cells. First, a range of lipid second messengers are formed from cell membrane phospholipids, including lyso-phospholipids, diacylglycerols and endocannabinoids. The fatty acid composition of these lipid second messengers affects their biological potency. For example, older data showed that the fatty acid composition of diacylglycerol affected its ability to activate protein kinase C (see Miles and Calder(Reference Miles and Calder21) for references), while endocannabinoids containing different fatty acids have different anti-inflammatory potency (see Calder(Reference Calder22) for references). It is obvious that the fatty acid composition of the lipid second messengers is determined by that of their parent phospholipid. Second, PUFA released mainly from the sn-2 position of membrane phospholipids act as substrates for COX, lipoxygenase (LOX) and cytochrome P450 enzymes to produce lipid mediators (eicosanoids) active in inflammation including PG and LT (Fig. 2). In this regard, the n-6 PUFA arachidonic acid is the most common cell membrane PUFA and the most common substrate for COX, LOX and cytochrome P450 enzymes. Several mediators formed from arachidonic acid, including PGD2, PGE2, LTB4 and the other four-series LT, are well-described mediators and regulators of inflammation(Reference Lewis, Austen and Soberman23–Reference Kroetz and Zeldin25). They act through binding to specific receptors, usually G protein-coupled receptors, and their synthesis and action are targets for a range of non-specific and specific anti-inflammatory pharmaceuticals. There is evidence from studies in both experimental animals(Reference Peterson, Jeffery and Thies26) and human subjects(Reference Rees, Miles and Banerjee27) that the synthesis of eicosanoids from arachidonic acid is related to the amount of the fatty acid available in cell membranes.
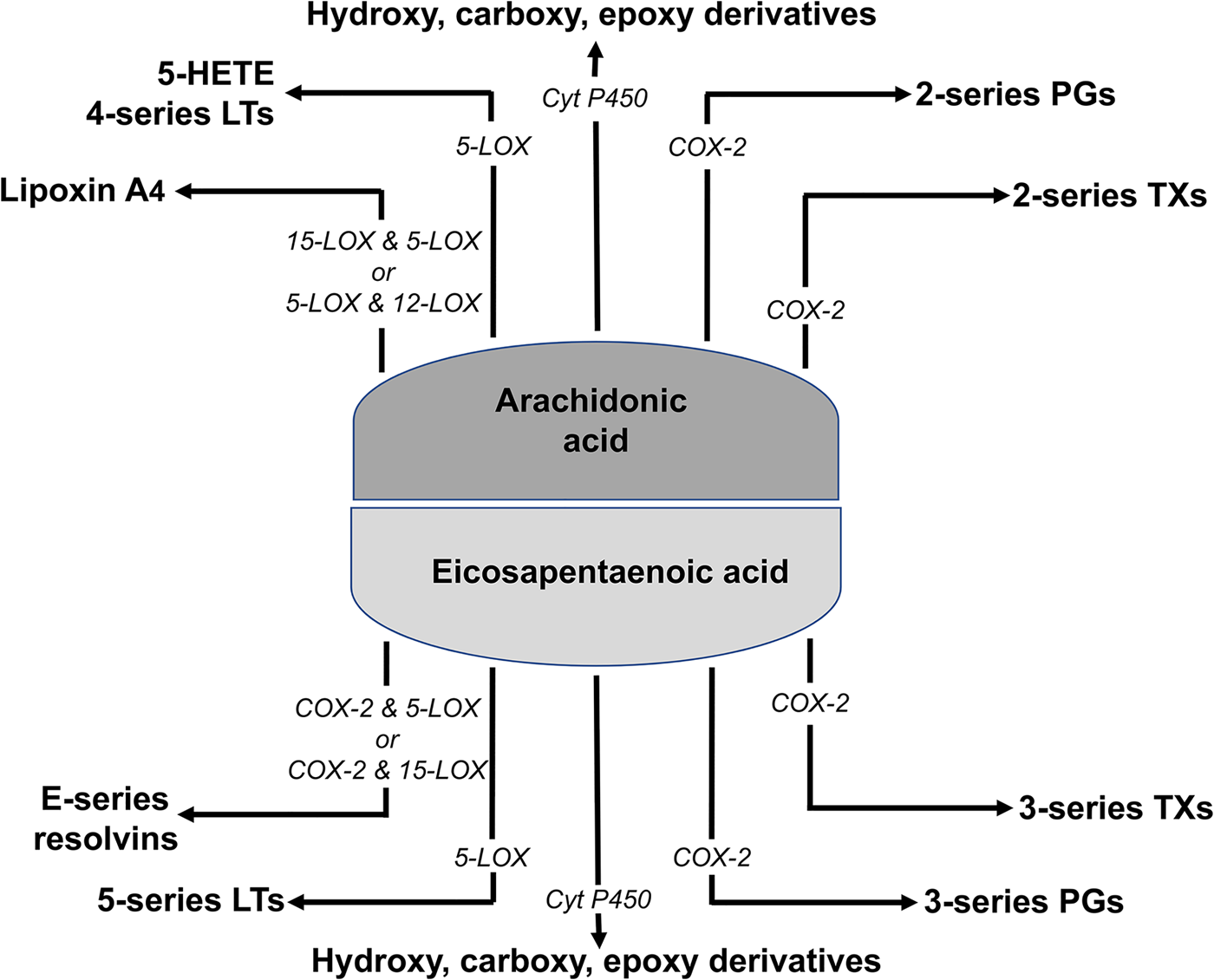
Fig. 2. (Colour online) Summary of eicosanoid synthesis from arachidonic and EPA. COX, cyclooxygenase; Cyt P450, cytochrome P450; HETE, hydroxyeicosatetraenoic acid; LOX, lipoxygenase; LT, leukotriene; TX, thromboxane.
The afore-mentioned considerations indicate that modification of the fatty acid composition of the membranes of cells involved in inflammation could alter inflammatory responses through altered NFκB activation, altered generation or potency of lipid second messengers and altered generation of eicosanoids; in this regard, there has been much interest in the effects of EPA and DHA. The phospholipids of blood cells involved in inflammatory processes (e.g. monocytes, lymphocytes (often studied together as mononuclear cells) and neutrophils) taken from human subjects consuming a typical Western diet typically contain 15–20 % of fatty acids as arachidonic acid, 0⋅5–1 % as EPA and 2–3 % as DHA(Reference Rees, Miles and Banerjee27–Reference Browning, Walker and Mander38). Increased intake of EPA and DHA results in increased amounts of EPA and DHA in these phospholipids(Reference Rees, Miles and Banerjee27–Reference Browning, Walker and Mander38). This enrichment in EPA and DHA occurs in a time-dependent(Reference Rees, Miles and Banerjee27,Reference Yaqoob, Pala and Cortina-Borja32–Reference Browning, Walker and Mander38) and a dose-dependent(Reference Rees, Miles and Banerjee27,Reference Kew, Banerjee and Minihane35,Reference Browning, Walker and Mander38) manner and is largely at the expense of arachidonic acid (Figs. 3 and 4). These changes in fatty acid composition seem to be important in modifying the production of lipid mediators and in regulating the formation of lipid rafts within membranes in response to an inflammatory stimulus (see later).

Fig. 3. Time-dependent changes in EPA and arachidonic acid content in human mononuclear cells. Healthy human participants consumed fish oil providing 2⋅1 g EPA and 1⋅1 g DHA daily for 1 week(Reference Faber, Berkhout and Vos37) or for 12 weeks(Reference Yaqoob, Pala and Cortina-Borja32). Participants in Faber et al.(Reference Faber, Berkhout and Vos37) were six females and six males while participants in Yaqoob et al.(Reference Yaqoob, Pala and Cortina-Borja32) were one female and seven males. Blood was sampled at several time points in each study and mononuclear cells prepared. Fatty acid composition of the cells was determined by GC. Mean values are shown. Squares represent EPA and triangles represent arachidonic acid. Black symbols represent data from Faber et al.(Reference Faber, Berkhout and Vos37) and grey symbols represent data from Yaqoob et al.(Reference Yaqoob, Pala and Cortina-Borja32). Reprinted from Biochimica et Biophysica Molecular and Cell Biology of Lipids, Vol 1851, P.C. Calder, Marine omega-3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance, pp. 469–484, Copyright 2015, with permission from Elsevier(Reference Calder22).

Fig. 4. Dose-dependent changes in EPA and arachidonic acid content in human mononuclear cells. Healthy males consumed a supplement providing 0, 1⋅35, 2⋅7 or 4⋅05 g EPA daily for 12 weeks (n 15 or 16). Blood was sampled at 0 and 12 weeks and mononuclear cells prepared. Fatty acid composition of the cells was determined by GC. Mean values for change from week 0 are shown; data for arachidonic acid have been normalised so that the change from week 0 in the group receiving no supplemental EPA is zero. Squares represent EPA and triangles represent arachidonic acid. Data are for the older males reported in Rees et al.(Reference Rees, Miles and Banerjee27) Reprinted from Biochimica et Biophysica Molecular and Cell Biology of Lipids, Vol 1851, P.C. Calder, Marine omega-3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance, pp. 469–484, Copyright 2015, with permission from Elsevier(Reference Calder22).
n-3 PUFA and eicosanoids
Since increased intake of EPA and DHA decreases the amount of arachidonic acid in the membrane phospholipids of cells involved in inflammation (Figs. 3 and 4), it is likely that the production of arachidonic acid-derived mediators would be decreased simply because of a reduced amount of available substrate. In addition, EPA and DHA have been shown to inhibit arachidonic acid metabolism and to decrease the expression of COX-2 gene and protein(Reference Lee, Kim and Chang39,Reference Baker, Valenzuela and De Souza40) . In accordance with these arguments, a number of studies in healthy human participants have described decreased production of two-series PG and four-series LT by inflammatory cells following the use of EPA and DHA in supplements for a period of weeks to months(Reference Rees, Miles and Banerjee27–Reference Caughey, Mantzioris and Gibson31,Reference Meydani, Endres and Woods41–Reference Trebble, Wootton and Miles43) . Similar effects have been described in patients with chronic inflammatory diseases such as RA(Reference Kremer, Bigauoette and Michalek44–Reference Tullekan, Limburg and Muskiet48) and inflammatory bowel disease(Reference McCall, O'Leary and Bloomfield49–Reference Trebble, Arden and Wootton53). These studies have often used quite high doses of EPA and DHA. Data from a dose–response study in healthy participants with an EPA-rich supplement showed that an EPA intake of 1⋅35 g/d for 3 months was not sufficient to influence ex vivo PGE2 production by LPS-stimulated mononuclear cells, whereas an EPA intake of 2⋅7 g/d significantly decreased PGE2 production(Reference Rees, Miles and Banerjee27). This study suggests a threshold for an anti-inflammatory effect of EPA of somewhere between 1⋅35 and 2⋅7 g EPA daily.
Like arachidonic acid, EPA is a substrate for the COX, LOX and cytochrome P450 enzymes that produce eicosanoids (Fig. 2). However, because of the structural differences between EPA and arachidonic acid, they generate eicosanoids with different structures, typically containing an additional double bond. Whereas arachidonic acid gives rise to two-series PG and four-series LT, EPA gives rise to three-series PG and five-series LT. Increased generation of five-series LT has been demonstrated with neutrophils from human subjects taking EPA and DHA supplements for several weeks(Reference Lee, Hoover and Williams28–Reference Sperling, Benincaso and Knoell30). Eicosanoids produced from EPA are usually less biologically potent than those produced from arachidonic acid. For example, LTB5 is 10- to 100-fold less potent than LTB4 as a leucocyte chemoattractant(Reference Goldman, Pickett and Goetzl54–Reference Bagga, Wang and Farias-Eisner56). One reason for this reduced biological potency is that eicosanoid receptors typically have a lower affinity for the EPA-derived mediator as described by Wada et al.(Reference Wada, DeLong and Hong57). For example, PGE3 had 20–50 % of the affinity of PGE2 for the EP1, EP2, EP3 and EP4 receptors. Thus, in general, it appears that EPA results in decreased production of potent eicosanoids from arachidonic acid and increased production of generally weak eicosanoids.
n-3 PUFA, lipid rafts, NFκB activation and inflammatory cytokines
As mentioned earlier, NFκB is one of the main transcription factors involved in up-regulation of the genes encoding proteins involved in inflammation including many cytokines, chemokines, adhesion molecules and COX-2(Reference Kumar, Takada and Boriek20,Reference Siga58) . Inactive NFκB is a cytosolic trimer, with one of the subunits being the so-called inhibitory subunit of NFκB (IκB). NFκB is activated through signalling cascades initiated by various extracellular inflammatory stimuli, including LPS binding to TLR4. In response to such stimuli, IκB is phosphorylated and dissociates from the remaining dimer; the dissociated IκB is degraded. The active dimeric NFκB translocates to the nucleus and binds to its DNA response elements up-regulating inflammatory gene expression(Reference Perkins59). EPA and fish oil decreased LPS-induced activation of NFκB in isolated monocytes(Reference Lo, Chiu and Fu60–Reference Novak, Babcock and Jho62), which was associated with decreased phosphorylation of IκB(Reference Novak, Babcock and Jho62,Reference Zhao, Joshi-Barve and Chen63) . Similarly, DHA reduced NFκB activation in response to LPS in cultured macrophages(Reference Lee, Sohn and Rhee64) and dendritic cells(Reference Weatherill, Lee and Zhao65,Reference Kong, Yen and Vassiliou66) ; again this involved decreased phosphorylation of IκB(Reference Lee, Sohn and Rhee64). This suggests an effect of EPA and DHA upstream of IκB phosphorylation. In contrast to these effects of EPA and DHA, some SFA, but particularly lauric acid, were able to increase the activation of NFκB and induce COX-2 expression in macrophages(Reference Lee, Sohn and Rhee64) and dendritic cells(Reference Weatherill, Lee and Zhao65). Lauric acid was not able to activate NFκB or induce COX-2 expression in macrophages that did not express TLR4(Reference Lee, Sohn and Rhee64). This suggests a direct interaction between lauric acid and TLR4. Similar to their effects in LPS-stimulated macrophages, both EPA and DHA were able to prevent the lauric acid-induced activation of NFκB and expression of COX-2(Reference Lee, Sohn and Rhee64). MyD88 is a cell membrane-associated adapter protein used by TLR4 in the early stages of the signalling cascade that eventually activates NFκB(Reference Ruysschaert and Lonez19). DHA did not inhibit COX-2 expression in macrophages not bearing constitutively active MyD88(Reference Lee, Sohn and Rhee64), which indicates that DHA inhibits LPS and lauric acid activation of NFκB upstream of MyD88. When inflammatory cells are stimulated by LPS, TLR4, MyD88 and other signalling proteins associate into lipid rafts(Reference Ruysschaert and Lonez19). Lauric acid was shown to induce this same raft formation in macrophages(Reference Wong, Kwon and Choi67). Furthermore, DHA inhibited the ability of both LPS and lauric acid to promote the recruitment of signalling proteins, including MyD88, into rafts(Reference Wong, Kwon and Choi67). These studies demonstrate two key points. First, the differential effects of lauric acid and n-3 PUFA on TLR4-induced inflammatory signalling that alter the activation of NFκB seem to be linked to the ability of those fatty acids to promote or to disrupt raft formation within the membrane of inflammatory cells. Second, the well-described effects of EPA and DHA on inflammatory gene (and protein) expression may actually be caused by very early events occurring in the cell membrane (Fig. 5).

Fig. 5. (Colour online) Overview of the key anti-inflammatory actions of EPA and DHA. TLR, Toll-like receptor.
One of the key actions of NFκB is to up-regulate the expression of inflammatory genes, including those encoding a number of cytokines such as TNF and several IL. Higher than normal levels of TNF, IL-1β, IL-6 and IL-8 are a common feature of many inflammatory conditions(Reference Calder, Albers and Antoine1,Reference Calder, Ahluwalia and Albers2) and also occur in obesity(Reference Calder, Ahluwalia and Brouns15) and with ageing(Reference Calder, Bosco and Bourdet-Sicard16). Effects of EPA and DHA on NFκB activation would be expected to result in decreased production of these cytokines. Indeed, EPA and DHA decreased LPS-stimulated production of IL-6 and IL-8 by cultured human endothelial cells(Reference de Caterina, Cybulsky and Clinton68,Reference Khalfoun, Thibault and Watier69) , and EPA and fish oil decreased LPS-induced TNF production by cultured monocytes(Reference Lo, Chiu and Fu60–Reference Novak, Babcock and Jho62). Both EPA and DHA decreased TNF-induced production of IL-6, IL-8, monocyte chemoattractant protein 1 and ‘regulated upon activation, normal T cell expressed and presumably secreted’ by cultured human endothelial cells, with DHA being more potent(Reference Baker, Valenzuela and De Souza40). Feeding fish oil to mice decreased the production of TNF, IL-1β and IL-6 by LPS-stimulated macrophages(Reference Wallace, Miles and Calder70–Reference Renier, Skamene and de Sanctis73) and decreased the blood concentrations of TNF, IL-1β and IL-6 following intraperitoneal injection of LPS(Reference Sadeghi, Wallace and Calder74). Fish oil is also reported to increase the concentration of the anti-inflammatory cytokine IL-10(Reference Sierra, Lara-Villoslada and Comalada75). Several studies providing EPA and DHA supplements to healthy human participants have reported decreased production of TNF, IL-1β and IL-6 by LPS-stimulated monocytes or mononuclear cells(Reference Endres, Ghorbani and Kelley29,Reference Caughey, Mantzioris and Gibson31,Reference Meydani, Endres and Woods41,Reference Trebble, Wootton and Miles43) , although not all studies report this effect, possibly because the dose of n-3 PUFA used was too low, although there may be other factors involved such as genotypic differences in responsiveness to n-3 fatty acids(Reference Grimble, Howell and O'Reilly76).
n-3 PUFA and PPAR-γ
PPAR-γ is a transcription factor which has anti-inflammatory effects(Reference Szanto and Nagy77). Agonists of PPAR-γ reduce murine colitis(Reference Desreumaux, Dubuquoy and Nutten78–Reference Dubuquoy, Rousseaux and Thuru80) and mice with PPAR-γ knock-down show enhanced susceptibility to chemically-induced colitis(Reference Desreumaux, Dubuquoy and Nutten78). PPAR-γ is able to physically interfere with the translocation of NFκB to the nucleus(Reference Berghe, Vermeulen and Delerive81). EPA and DHA can activate PPAR-γ(Reference Forman, Chen and Evans82–Reference Krey, Braissant and L'Horset85). Furthermore, DHA induced PPAR-γ in dendritic cells, which was associated with inhibition of NFκB activation and reduced production of TNF and IL-6 following LPS stimulation(Reference Kong, Yen and Vassiliou66). DHA also induced a number of PPAR-γ target genes in dendritic cells(Reference Zapata-Gonzalez, Rueda and Petriz86). The EPA derivatives PGD3 and 15-deoxy-PGD3 activated PPAR-γ in adipocytes, which is linked to the induction of the anti-inflammatory adipokine adiponectin(Reference Lefils-Lacourtablaise, Socorro and Géloën87). These observations suggest that one mechanism of the anti-inflammatory action of EPA and DHA is the activation of PPAR-γ. This may be another means through which these fatty acids inhibit NFκB activation (Fig. 5).
n-3 PUFA and specialised pro-resolving mediators
Both EPA and DHA are substrates for the synthesis of SPM (Fig. 6). SPM include the E- and D-series resolvins produced from EPA and DHA, respectively, and protectins (aka neuroprotectins) and maresins produced from DHA. Generation of classic eicosanoids involves either the COX or LOX pathways operating separately from one another (Fig. 2). However, SPM are synthesised using COX and LOX enzymes in the same pathway (Fig. 6). Furthermore, synthesis of many SPM is promoted by aspirin and different epimers are produced in the presence and absence of aspirin(Reference Bannenberg and Serhan88–Reference Serhan, Chiang and Dalli90). Both aspirin-triggered and non-aspirin-triggered SPM have biological activity(Reference Bannenberg and Serhan88–Reference Serhan, Chiang and Dalli90). A variety of SPM have been reported in human blood(Reference Colas, Shinohara and Dalli91–Reference Barden, Mas and Croft93), including umbilical cord blood(Reference See, Mas and Prescott94,Reference Nordgren, Berry and van Ormer95) , adipose tissue(Reference Titos, Rius and Lopez-Vicario96), breast milk(Reference Weiss, Troxler and Klinke97) and synovial fluid(Reference Sano, Toyoshia and Miki98). Supplemental EPA and DHA has been shown to result in higher concentrations of some SPM in human blood(Reference Colas, Shinohara and Dalli91–Reference Barden, Mas and Croft93,Reference Polus, Zapala and Razny99) . Ostermann et al.(Reference Ostermann, West and Schoenfeld100) demonstrated a dose-dependent increase in the plasma concentrations of multiple precursors of SPM in healthy participants supplemented with different doses of EPA + DHA for 1 year (Fig. 7), although the SPM themselves were not detected. Maternal supplementation with high-dose fish oil during pregnancy resulted in higher concentrations of the SPM precursors 17-hydroxy DHA and 18-hydroxy EPA, but not of the SPM themselves, in umbilical cord blood(Reference See, Mas and Prescott94). Patients with peripheral artery disease showed increased plasma resolvin E3 after 3 months supplementation with high-dose EPA + DHA(Reference Ramirez, Gasper and Khetani101). In healthy participants, single dosing with an SPM-enriched fish oil resulted in a dose-dependent elevation in plasma SPM concentrations over the following hours(Reference Souza, Marques and Gomez102).
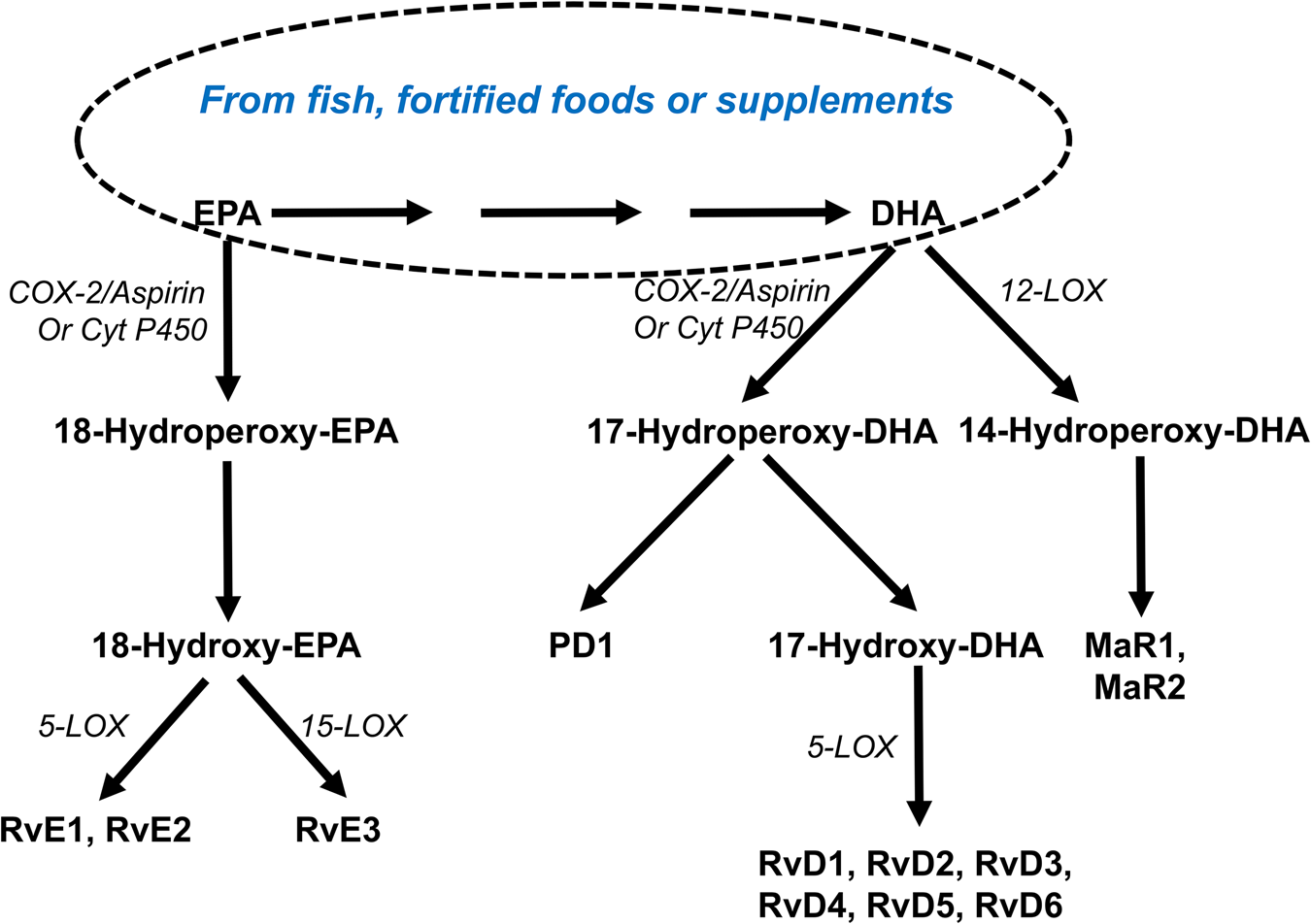
Fig. 6. (Colour online) Overview of the synthesis of specialised pro-resolving lipid mediators from EPA and DHA. COX, cyclooxygenase; Cyt P450, cytochrome P450 enzymes; LOX, lipoxygenase; MaR, maresin; P, protectin; Rv, resolvin.

Fig. 7. Plasma concentrations of three precursors of specialised pro-resolving mediators in human subjects. Healthy human participants consumed fish oil providing different amounts of EPA and DHA per week for 1 year. The different groups comprised fourteen males and fourteen females (placebo), seventeen males and eighteen females (3⋅27 g EPA + DHA/week), fourteen males and fifteen females (6⋅54 g EPA + DHA per week) and fifteen males and fourteen females (13⋅08 g EPA + DHA/week). Lipid mediator concentrations were determined by liquid chromatography-MS. Circles represent 8-hydroxy-EPA; squares represent 14-hydroxy-DHA; triangles represent 17-hydroxy-DHA. Data are taken from Ostermann et al.(Reference Ostermann, West and Schoenfeld100).
The biological effects of SPM have been examined extensively in cell culture and animal models of inflammation(Reference Bannenberg and Serhan88–Reference Serhan, Chiang and Dalli90) and are now beginning to be explored in human subjects. The cell and animal models have shown that all SPM tested to date have anti-inflammatory and inflammation-resolving actions. For example, resolvin E1, resolvin D1 and protectin D1 all inhibited transendothelial migration of neutrophils, so preventing the infiltration of neutrophils into sites of inflammation; resolvin D1 inhibited IL-1β production; and protectin D1 inhibited TNF and IL-1β production(Reference Bannenberg and Serhan88–Reference Serhan, Chiang and Dalli90). Resolvins reduce inflammation and protect experimental animals in models of inflammatory disease including arthritis(Reference Lima-Garcia, Dutra and da Silva103,Reference Benabdoun, Kulbay and Rondon104) , colitis(Reference Arita, Yoshida and Hong105), asthma(Reference Aoki, Hisada and Ishizuka106–Reference Rogerio, Haworth and Croze109) and other states of inflammation including sepsis(Reference Spite, Norling and Summers110,Reference Chen, Fan and Wu111) and acute lung injury(Reference Seki, Fukunaga and Arita112–Reference Liao, Dong and Wu114). The potent activity of SPM may explain many of the documented actions of EPA and DHA in inflammation.
n-3 PUFA and rheumatoid arthritis
RA is a chronic inflammatory disease that affects the joints, with infiltration of inflammatory cells(Reference Sweeney and Firestein115), increased expression of both COX-1 and COX-2 in the synovium along with high concentrations of pro-inflammatory eicosanoid products of arachidonic acid metabolism (e.g. PGE2) in the synovial fluid(Reference Sano, Hla and Maier116) and high concentrations of pro-inflammatory cytokines including TNF, IL-1β, IL-6, IL-8 and granulocyte/macrophage colony-stimulating factor in the synovial fluid and circulation(Reference Feldmann and Maini117). These observations indicate that RA may be a target for EPA + DHA. Mice fed fish oil had delayed onset and reduced incidence and severity of collagen-induced arthritis compared to mice fed vegetable oil(Reference Leslie, Gonnerman and Ullman118). EPA and DHA both suppressed streptococcal cell wall-induced arthritis in rats, with EPA being more effective(Reference Volker, FitzGerald and Garg119). Both fish oil and krill oil slowed the onset of collagen-induced arthritis in mice, decreasing its severity, paw swelling and knee joint pathology compared with the control group(Reference Ierna, Kerr and Scales120). EPA and DHA supplements lowered blood concentrations of inflammatory cytokines(Reference Kremer, Lawrence and Jubiz121–Reference Kremer, Lawrence and Petrillo124) and eicosanoids(Reference Cleland, French and Betts46,Reference Sperling, Weinblatt and Robin125,Reference van der Tempel, Tullekan and Limburg126) in patients with RA. In a recent trial, DHA at 2⋅1 g/d for 10 weeks increased plasma concentrations of SPM precursors (14- and 17-hydroxy DHA) in patients with RA(Reference Dawczynski, Dittrich and Neumann127). These effects should reduce pain and cartilage destruction; if pain is reduced then patients may decrease their use of pain-controlling drugs such as non-steroidal anti-inflammatory drugs (NSAID). In agreement with this, Cleland et al.(Reference Cleland, Caughey and James47) reported that patients with RA who used fish oil supplements were more likely to reduce the use of NSAID and to be in remission than those patients who did not use fish oil. A number of randomised controlled trials of fish oil in RA report improvements in several clinical outcomes including reduced duration of morning stiffness, reduced number of tender or swollen joints, reduced joint pain, reduced time to fatigue, increased grip strength and decreased use of NSAID (see(Reference Calder128,Reference Miles and Calder129) for references). The dose of EPA + DHA used in these trials has usually been quite high, between about 1 and 7 g daily, averaging about 3⋅5 g daily. In keeping with the difference in the prevalence of RA between males and females, most of these studies recruited many more females than males; no trials have investigated or compared the effect of n-3 fatty acids in females and males with RA. Several systematic reviews and meta-analyses of trials of fish oil in patients with RA have been conducted(Reference Fortin, Lew and Liang130–Reference Abdulrazaq, Innes and Calder133). One meta-analysis included data from nine trials published between 1985 and 1992 inclusive and from one unpublished trial and concluded that dietary fish oil supplementation for 3 months significantly reduced tender joint count and morning stiffness(Reference Fortin, Lew and Liang130). A second meta-analysis of n-3 fatty acids and pain included data from seventeen trials(Reference Goldberg and Katz131); this analysis indicated that fish oil reduces patient assessed joint pain, duration of morning stiffness, number of painful and/or tender joints, and use of NSAID. In 2017, a systematic review(Reference Senftleber, Nielsen and Andersen132) and a systematic review and meta-analysis(Reference Abdulrazaq, Innes and Calder133) of n-3 PUFA supplements and arthritic pain were published. The first of these included eighteen randomised controlled trials that used between 2⋅1 and 9⋅1 g EPA + DHA/d for durations of 12–52 weeks. Ten of these studies supported the hypothesis that n-3 PUFA reduce patient or physician assessment of pain in patients with RA(Reference Senftleber, Nielsen and Andersen132). The meta-analysis of twenty-two trials identified a significant reduction in pain with n-3 PUFA in patients with RA(Reference Abdulrazaq, Innes and Calder133). Thus, based upon the findings of individual trials and meta-analyses of those findings, there is fairly robust evidence of the efficacy of n-3 fatty acids in RA, although high doses seem to be needed. A recent trial using foods enriched with algal oil providing 2⋅1 g DHA daily for 10 weeks in RA patients maintaining their medication regimen found a significant reduction in tender and swollen joints, disease activity score and ultrasound score(Reference Dawczynski, Dittrich and Neumann127).
One study has examined the relationship between EPA status and responsiveness of patients with RA to treatment with antibodies against TNF(Reference Jeffery, Fisk and Calder134). It was identified that the greatest reduction in disease activity score at 12 weeks of treatment was seen in those in the highest tertile of plasma phospholipid EPA. Furthermore, plasma phospholipid EPA was significantly positively associated with European league against rheumatism response. These observations indicate that response to anti-TNF antibodies is better in those with higher EPA status. An increase in Th17 cells has been associated with non-response to anti-TNF antibodies. In vitro incubation of blood mononuclear cells with the antibodies increased the frequency of Th17 cells, but co-incubation with EPA prevented this(Reference Jeffery, Fisk and Calder134). It was suggested that increasing EPA status might improve the response of patients with RA to anti-TNF antibodies by preventing the generation of pro-inflammatory Th17 cells.
Decreased inflammation with n-3 PUFA: any concern about infection?
Because inflammation is the earliest stage of the host response to infection, whether the anti-inflammatory effects of n-3 PUFA increase susceptibility to infection requires consideration. This is discussed in some detail elsewhere(Reference Calder135–Reference Husson, Ley and Portal137), although the focus has been mainly on studies in mice, rats and guinea pigs. The conclusion of the most recent discussion(Reference Husson, Ley and Portal137), based on these animal studies, is that low-dose EPA + DHA is beneficial against experimental infections caused by extracellular pathogens (which induce a strong inflammatory response) including Staphylococcus pneumoniae, Pseudomonas aeruginosa, Escherichia coli, and Staphylococcus aureus by reducing inflammation. A concern was raised about higher doses of EPA + DHA and infections by intracellular pathogens such as Mycobacterium tuberculosis, Salmonella, influenza virus and Herpes simplex virus(Reference Husson, Ley and Portal137). Since these conclusions are based on animal studies, where the extrapolation of dietary intakes to human subjects is uncertain, and where there are also other dietary differences as well as differences in gut microbiota, lipid metabolism and the immune system, they must be treated cautiously. Follow-up over 10 years of 38 378 male US health professionals aged 44–79 years at study outset identified no significant relationship between EPA or DHA intake and risk of community-acquired pneumonia(Reference Merchant, Curham and Rimm138). In a cohort of 63 257 Chinese men and women aged 45–74 years recruited between 1993 and 1998, intake of EPA + DHA was associated with a reduced risk of developing active tuberculosis in the follow-up period to 2013 in a dose-dependent manner(Reference Soh, Chee and Wang139). Two intervention studies in children indicate that EPA + DHA do not increase, and may even decrease, the risk of infections. Thai schoolchildren aged 9–12 years consuming milk fortified with fish oil providing 200 mg EPA + 1 g DHA daily on 5 d per week for 6 months had significantly fewer episodes and shorter duration of illness (mainly upper respiratory tract) than the placebo group(Reference Thienprasert, Samuhaseneetoo and Popplestone140). Iron-deficient South African schoolchildren aged 6–11 years received iron, EPA + DHA (80 mg EPA + 420 mg DHA) or iron + EPA + DHA four times per week for 8⋅5 months(Reference Malan, Baumgartner and Calder141). Iron supplementation increased the number of days with illness and illness caused by respiratory symptoms, whereas EPA + DHA reduced the number of days with illness at school. Furthermore, the combination of EPA + DHA with iron prevented the adverse effect of iron alone. Studies of n-3 PUFA and infection in adults have mainly been studied in hospitalised patients. Intravenous administration of lipid emulsions containing fish oil to surgical patients has been reported to reduce infections in a number of trials; as discussed elsewhere(Reference Calder142), six meta-analyses published between 2010 and 2018 all report significantly reduced risk of infection with odds/risk ratios of between 0⋅36 and 0⋅56 compared with placebo. In critically ill patients, the evidence for intravenous fish oil is less clear(Reference Calder142), but the most recent meta-analysis(Reference Manzanares, Langlois and Dhaliwal143) identified a risk ratio of 0⋅64 for infections in patients receiving intravenous fish oil compared to placebo. Enteral feeds containing fish oil in combination with other bioactive nutrients have been demonstrated through meta-analysis to reduce infections in surgical(Reference Marik and Zaloga144) and critically ill(Reference Marik and Zaloga145) patients.
Conclusions
Inflammation is a normal part of the immune response and should be self-limiting. It involves a multitude of cell types, chemical mediators and interactions. Excessive or unresolved inflammation is linked to tissue damage, pathology and many conditions of ill health. Chemical mediators produced from PUFA are known to play a role in the initiation, perpetuation and termination of inflammatory responses and changes in fatty acid composition can modify lipid raft formation and cell signalling leading to altered gene expression and an altered pattern of lipid mediator production. Cells involved in the inflammatory response are typically rich in the n-6 PUFA arachidonic acid, but the contents of arachidonic acid and of EPA and DHA can be altered through oral administration of EPA and DHA. Eicosanoids produced from arachidonic acid, such as PGE2 and four-series LT, have roles in inflammation. EPA also gives rise to eicosanoids but these are usually less potent than those produced from arachidonic acid. EPA and DHA give rise to resolvins, and DHA to protectins and maresins which are anti-inflammatory and inflammation resolving. The effects of EPA and DHA on inflammatory gene expression are due at least in part to reduced activation of NFκB which seems to relate to membrane-mediated events including inhibition of lipid raft formation in response to inflammatory triggers. Dose-dependent actions of n-3 PUFA on inflammatory responses have not been well described, but it appears that a dose of at least 2 g daily is necessary to achieve an anti-inflammatory effect in human subjects. As a result of their anti-inflammatory actions, EPA and DHA may have therapeutic efficacy in inflammatory diseases. Work with animal models of RA has demonstrated the efficacy of fish oil and of mediators derived from EPA and DHA, such as some of the resolvins. There have been a number of clinical trials of fish oil in patients with RA and these trials have typically used high doses of EPA + DHA, often above the anti-inflammatory threshold of 2 g daily. Many trials in RA report clinical improvements (e.g. improved patient assessed pain, decreased morning stiffness, fewer painful or tender joints, decreased use of NSAID), and when the trials have been pooled in meta-analyses, statistically significant clinical benefit has emerged(Reference Fortin, Lew and Liang130–Reference Abdulrazaq, Innes and Calder133). Thus, based upon the findings of individual trials and meta-analyses of those findings, there is fairly robust evidence of the efficacy of n-3 fatty acids in RA, although high doses seem to be needed. Decreasing inflammation with n-3 PUFA appears not to be associated with an impairment of host defence where that has been tested in human subjects, and may even enhance protection. Trials of EPA + DHA and infection in adults in the community (i.e. non-hospitalised) are warranted.
Financial Support
None.
Conflict of Interest
P. C. C. has received research funding from BASF AS and acts as a consultant to BASF AS, Smartfish, DSM, Cargill and Fresenius-Kabi.
Authorship
The author had sole responsibility for all aspects of preparation of this paper.







