



Introduction
Arsenic mineralogy is a very interesting topic, owing to the complex crystal-chemistry of this element, occurring in more than 400 mineral species, and its environmental significance (e.g. Majzlan et al., Reference Majzlan, Drahota, Filippi, Bowell, Alpers, Jamieson, Nordstrom and Majzlan2014). Usually, arsenic occurs in arsenates as As5+, forming the oxy-anion (AsΦ4), where Φ is a ligand [e.g. O2–, (OH)–…]; the oxy-anion (As3+Φ3) is definitely rarer, occurring in only a few minerals. As the Pauling's bond strength for each bond in an (As3+Φ3) group is 1 valence unit (vu), such groups can be polymerised, giving rise to different possible configurations. Consequently, in addition to isolated (As3+Φ3) groups, (As2O5) dimers, (As4O8) tetramers and (As5O11) pentamers are known in minerals. In addition, infinite chains have been observed.
Tetramers, pentamers and infinite chains are rare, having been found in only four mineral species. Rings formed by four (AsO3) groups occur in stenhuggarite, ideally CaFeSbAs2O7 (Coda et al., Reference Coda, Dal Negro, Sabelli and Tazzoli1977), whereas pentamers were observed in ludlockite, PbFe4As10O22 (Cooper and Hawthorne, Reference Cooper and Hawthorne1996). Infinite chains have been reported in trippkeite, CuAs2O4 (Pertlik, Reference Pertlik1975) and leiteite, ZnAs2O4 (Ghose et al., Reference Ghose, Gupta and Schlemper1987). Species containing (As2O5) dimers are relatively more common. Table 1 lists the minerals characterised by this structural feature. In schneiderhöhnite, Fe2+Fe3+3As5O13, (As2O5) dimers occur along with isolated (AsO3) groups (Hawthorne, Reference Hawthorne1985; Cooper and Hawthorne, Reference Cooper and Hawthorne2016), whereas karibibite displays (As2O5) dimers and (AsO2) chains (Colombo et al., Reference Colombo, Mugnaioli, Vallcorba, García, Goñi and Rius2017).
Table 1. Minerals characterised by the occurrence of (As2O5) dimers.

During the investigation of the mineralogy of the Monte Arsiccio mine in the Apuan Alps, northern Tuscany, Italy, the arsenite mineral graeserite was identified in veins hosted in metadolostone (Biagioni et al., Reference Biagioni, Bonaccorsi, Perchiazzi, Hålenius and Zaccarini2020a). This mineral is sometimes associated with square tabular crystals, usually deeply altered to an earthy X-ray amorphous compound. Some relicts of the original mineral were eventually found at the core of one of these altered crystals and some unaltered individuals were collected. This made it possible to obtain some material for the mineralogical investigation that led to recognition of this phase as a new mineral species, bianchiniite. The mineral and its name have been approved by the Commission on New Minerals, Nomenclature and Classification (CNMNC) of the International Mineralogical Association (IMA) under the number IMA2019-022 (Biagioni et al., Reference Biagioni, Pasero, Hålenius and Bosi2019c). The holotype material, represented by the grain used for the collection of all the mineralogical data, is deposited in the collection of the Museo di Storia Naturale, Università di Pisa, Via Roma 79, Calci, Pisa, Italy, with catalogue number 19892. The name honours the mineral collectors Andrea Bianchini (born 5th September 1959) and Mario Bianchini (born 28th August 1962) (no relationship between them) for their contribution to the knowledge of the mineralogy of pyrite ± baryte ± iron-oxide ore deposits of the southern Apuan Alps, northern Tuscany, Italy. Andrea Bianchini provided the first specimens of dessauite-(Y) (Orlandi et al., Reference Orlandi, Pasero, Duchi and Olmi1997), as well as other rare mineral species from the Buca della Vena mine. At the beginning of the 1990s, he was the first to collect samples of a Ti–Fe arsenite/antimonite mineral from the Monte Arsiccio mine; several years later, these samples were identified as Sb-rich graeserite (Biagioni et al., Reference Biagioni, Bonaccorsi, Perchiazzi, Hålenius and Zaccarini2020a). Mario Bianchini provided several mineral samples for the investigation of the mineralogy of ore deposits located in the Sant'Anna tectonic window (in particular, the Pollone and Monte Arsiccio mines). He found the first specimens of the new mineral species giacovazzoite (Biagioni et al., Reference Biagioni, Bindi, Mauro and Pasero2020b), magnanelliite (Biagioni et al., Reference Biagioni, Bindi and Kampf2019a) and scordariite (Biagioni et al., Reference Biagioni, Bindi, Mauro and Hålenius2019b), as well as the only available material of bianchiniite (along with the mineral collector Stefano Magnanelli).
The aim of this paper is to describe this new arsenite mineral and discuss its intriguing crystal chemistry.
Occurrence and mineral description
Occurrence and physical properties
Bianchiniite was found in only a few samples collected in the Sant'Olga level, Monte Arsiccio mine (43°58′N, 10°17′E), near Sant'Anna di Stazzema, Stazzema, Apuan Alps, Tuscany, Italy. The Monte Arsiccio mine exploited a pyrite ± baryte ± iron-oxide (magnetite, hematite and ‘limonite’) ore deposit located in the north-eastern sector of the Sant'Anna tectonic window, in the southern Apuan Alps. A review on its geological setting can be found in Biagioni et al. (Reference Biagioni, D'Orazio, Fulignati, George, Mauro and Zaccarini2020c) and references therein.
In type material, bianchiniite is associated with baryte, ‘hyalophane’ and ‘chlorite’, in a fracture of metadolostone (Fig. 1). After the first finding, which provided the material used for the type description, two additional specimens of bianchiniite were identified. The first one has the same associated minerals as observed in type material, plus galena in cuboctahedral crystals. Other phases identified in the same occurrence are sphalerite, derbylite, graeserite, siderite, dolomite, ankerite and quartz. Usually, crystals of bianchiniite are deeply altered to an X-ray amorphous earthy material (Fig. 1), in some cases admixed with microcrystalline rutile; in the second additional identified specimen, a relict of bianchiniite was observed at the core of a deeply altered crystal, confirming the identity of the original pseudomorphosed mineral. The crystallisation of bianchiniite is probably related to the circulation of As-rich hydrothermal fluids during the Alpine tectono-metamorphic event, under specific $f_{{\rm O}_ 2}$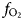 conditions, within the metadolostone rock body embedded in the Monte Arsiccio ore deposit. The alteration affecting the large majority of the observed crystals of bianchiniite may suggest a successive alteration by low-T fluids, during the final stages of the evolution of the Monte Arsiccio mineral assemblages.
conditions, within the metadolostone rock body embedded in the Monte Arsiccio ore deposit. The alteration affecting the large majority of the observed crystals of bianchiniite may suggest a successive alteration by low-T fluids, during the final stages of the evolution of the Monte Arsiccio mineral assemblages.

Fig. 1. (a) A loose aggregate of tabular crystals of bianchiniite, associated with ‘hyalophane’ and ‘chlorite’. Holotype material was sampled from this specimen. (b) Deeply altered earthy tabular crystals of bianchiniite associated with rhombohedral crystals of siderite and whitish microcrystalline fibrous aragonite. Both specimens from the Sant'Olga level, Monte Arsiccio mine, Stazzema, Apuan Alps, Tuscany, Italy (in a private collection).
Bianchiniite occurs as square tabular {001} crystals, up to 1 mm across. The colour is brown and the streak is brownish. The mineral is transparent, with a vitreous lustre. Owing to the small size of the grains available for the collection of mineralogical data, hardness was not measured. Bianchiniite is brittle, with a perfect {001} cleavage; fracture is irregular. Density was not measured; on the basis of the empirical formula and unit-cell volume refined from single-crystal X-ray diffraction data, the calculated density is 4.863 g/cm3.
The holotype material was embedded in epoxy and subsequently optical properties were determined on the polished crystal using reflected light microscopy. Reflectance was measured using an AVASPEC-UL2048×16 spectrometer attached to a Zeiss Axiotron UV-microscope (Swedish Museum of Natural History, Stockholm, Sweden), using a halogen lamp (100 W), with a measured field of 30 μm in diameter. In plane-polarised light, bianchiniite is grey in colour, non-pleochroic, with orange–yellow internal reflections. Between crossed polars, this mineral is weakly bireflectant, with a very weak anisotropy in shades of grey. Reflectance values (SiC as standard), measured in air, are given in Table 2. Mean refractive index, calculated according to the Gladstone–Dale relationship (Mandarino, Reference Mandarino1979, Reference Mandarino1981), is 2.044.
Table 2. Reflectance data (%) for bianchiniite in air.*
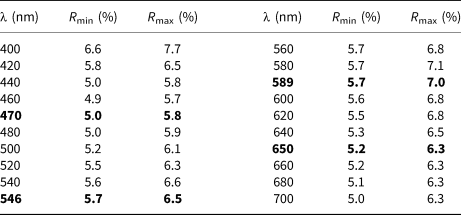
* The reference wavelengths required by the Commission on Ore Mineralogy (COM) are given in bold
Chemical and micro-Raman data
Quantitative chemical data were collected using a Cameca SX50 electron microprobe at the Istituto di Geologia Ambientale e Geoingegneria, CNR, Rome, Italy. The following analytical conditions were used: wavelength dispersive mode, accelerating voltage = 15 kV, beam current = 15 nA and beam size = 1 μm. Standards (element, emission line) were: rutile (TiKα), vanadinite (VKα), magnetite (FeKα), GaAs (AsLα), InSb (SbLα), celestine (SrLα), baryte (BaLα), galena (PbMα) and fluorophlogopite (FKα). Chromium and Cl were sought but were below the detection limit. Chemical data are given in Table 3. Vanadium, Fe and As were considered as V3+, Fe3+ and As3+, respectively, on the basis of the crystal structure refinement (see below). On the basis of 12 anions per formula unit, the empirical formula of bianchiniite is (Ba2.00Sr0.04Pb0.02)Σ2.06(Ti4+1.14V3+0.44Fe3+0.42)Σ2.00[(As3.96Sb0.02)Σ3.98O10](O1.18F0.82), corresponding to the end-member formula Ba2(Ti4+V3+)(As2O5)2OF, in accordance with the CNMNC-IMA guidelines (Bosi et al., Reference Bosi, Hatert, Hålenius, Pasero, Miyawaki and Mills2019). This latter formula corresponds to (in wt.%) TiO2 9.20, V2O3 8.63, As2O3 45.57, BaO 35.32, F 2.19, sum 100.92, –O = F –0.92, total 100.
Table 3. Electron microprobe data (in wt.% – average of 10 spot analyses) and atoms per formula unit for bianchiniite.
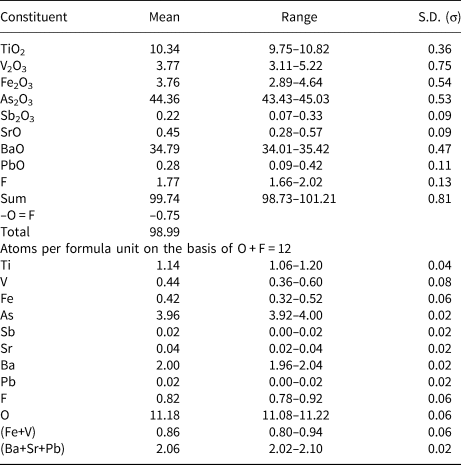
S.D. – standard deviation
Micro-Raman spectra of bianchiniite were collected in near back-scattered geometry with a Horiba Jobin-Yvon Xplora Plus apparatus, equipped with a motorised x–y stage and an Olympus BX41 microscope with a 100× objective (Dipartimento di Scienze della Terra, Università di Pisa). The 532 nm line of a solid-state laser was used. The minimum lateral and depth resolution was set to a few μm. The system was calibrated using the 520.6 cm–1 Raman band of Si before each experimental session. Spectra were collected through multiple acquisitions (3) with single counting times of 60 s, with the laser power filtered at 50% (i.e. 12.5 mW). No thermal damage was observed. Back-scattered radiation was analysed with a 1200 gr/mm grating monochromator. The Raman spectrum of bianchiniite in the region between 100 and 1200 cm–1 is shown in Fig. 2. The interpretation of the Raman bands is not straightforward, as very few Raman spectroscopic studies on diarsenites are available (e.g. Čejka et al., Reference Čejka, Bahfenne, Frost and Sejkora2008; Bahfenne et al., Reference Bahfenne, Rintoul, Langhof and Frost2012). Bands between 750 and 850 cm–1 may be related to As–Onbr stretching modes, where subscript ‘nbr’ indicates ‘non-bridging’ oxygen atoms in the (As2O5) groups, in agreement with Bahfenne et al. (Reference Bahfenne, Rintoul, Langhof and Frost2012) and the theoretical calculations of Tossell (Reference Tossell1997). In particular, the band at 766 cm–1 seems to be the most probable candidate for this assignment, as it is very close to the value observed in fetiasite (770 cm–1; Graeser et al., Reference Graeser, Schwander, Demartin, Gramaccioli, Pilati and Reusser1994) and interpreted by Tossell (Reference Tossell1997) as due to the As–Onbr stretching vibrations. Other bands in this range, as well as the bands at 892 cm–1 can be also related to other M–O modes, where M can be Ba or (Ti,V,Fe). Bands in the range between 480 and 660 cm–1 are attributed to the As–Obr stretching mode, where subscript ‘br’ indicates ‘bridging’ oxygen atoms. More questionable is the correct attribution of the strong band at 658 cm–1. Indeed, this band is similar to the one reported in the infrared spectrum of fetiasite (at 660 cm–1; Graeser et al., Reference Graeser, Schwander, Demartin, Gramaccioli, Pilati and Reusser1994) and interpreted by Tossell (Reference Tossell1997) as due to the As–Obr stretching mode. However, these values do not match with the experimental data given for paulmooreite, with bands at 562 and 503 cm–1, and with the theoretical ones reported by Tossell (Reference Tossell1997), who calculated stretching wavenumbers at 554 and 496 cm–1. Consequently, the strong band at 658 cm–1 could have another interpretation; for instance, it may be due to (Ti,V,Fe)–O vibrations. The band at 295 cm–1 may be due to deformations of the As–Onbr bonds (e.g. Bahfenne et al., Reference Bahfenne, Rintoul, Langhof and Frost2012). Other bands below 300 cm–1 can be probably attributed to lattice or M–O bonds. In paulmooreite, bands at 186 and 138 cm–1 were observed (Bahfenne et al., Reference Bahfenne, Rintoul, Langhof and Frost2012), comparable to those at 178 and 140 cm–1 measured in bianchiniite. No spectral features were observed in the range 2800–4000 cm–1, thus supporting the absence of (OH)– and H2O groups in bianchiniite.
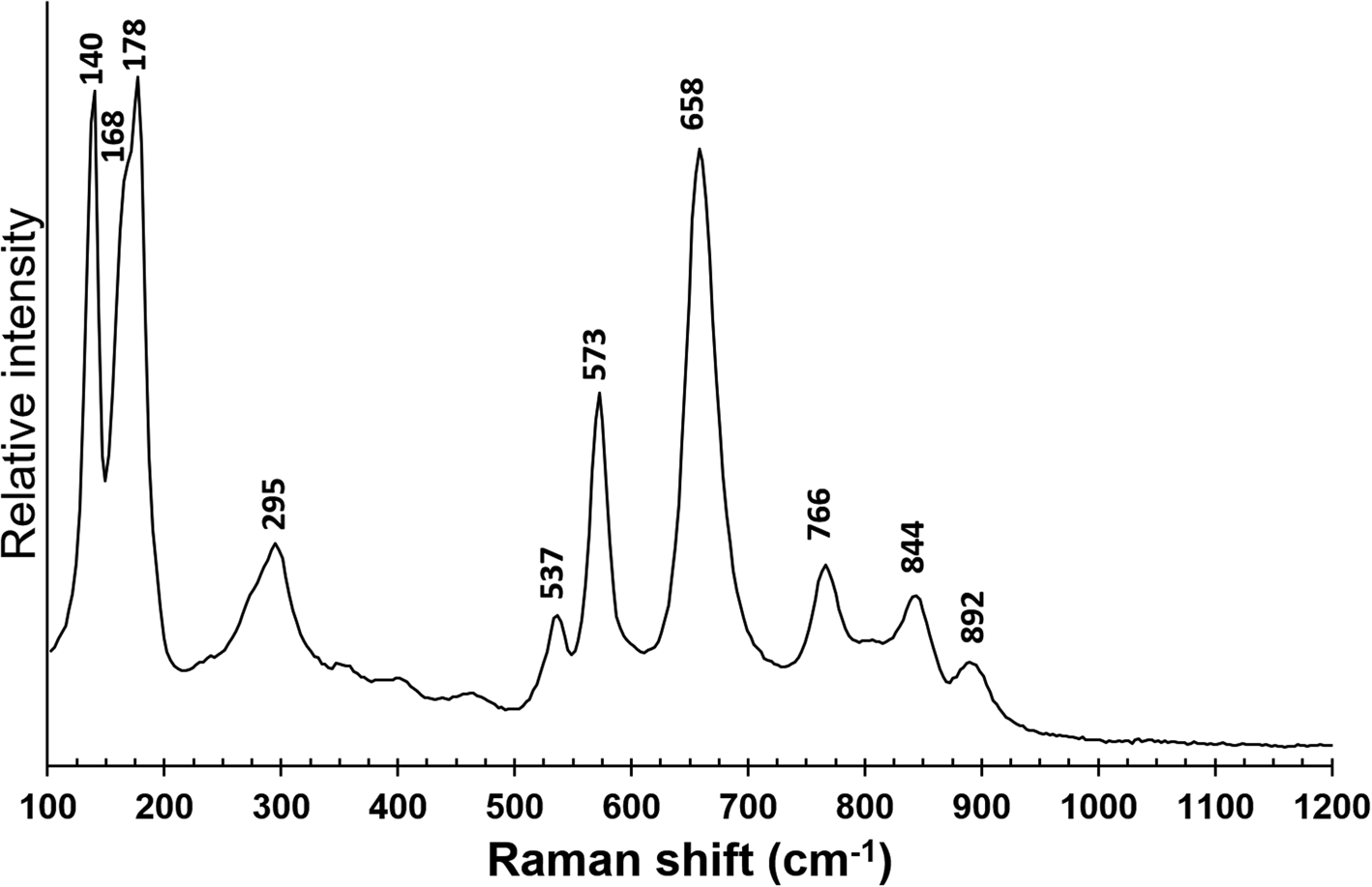
Fig. 2. Micro-Raman spectrum of bianchiniite in the range between 100 and 1200 cm–1.
Crystallography
Powder X-ray diffraction data of bianchiniite were collected using a 114.6 mm Gandolfi camera and Ni-filtered CuKα radiation (Dipartimento di Scienze della Terra, Università di Pisa). Table 4 gives the observed powder X-ray diffraction lines, along with the calculated pattern based on the structural model discussed below. Unit-cell parameters were refined in a tetragonal setting on the basis of 16 unequivocally indexed reflections using the software UnitCell (Holland and Redfern, Reference Holland and Redfern1997), giving the following results: a = 8.7894(10), c = 15.637(2) Å and V = 1208.0(2) Å3.
Table 4. X-ray powder diffraction data (d in Å) for bianchiniite.*
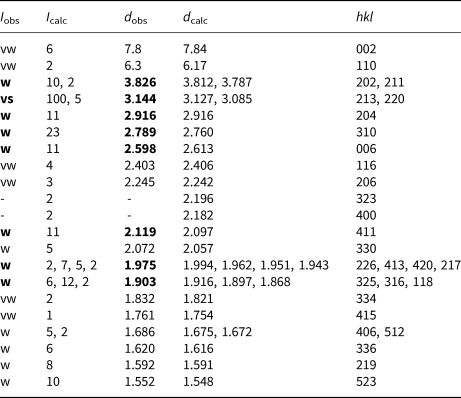
* Notes: intensity (I) and d hkl were calculated using the software PowderCell 2.3 (Kraus and Nolze, Reference Kraus and Nolze1996) on the basis of the structural model given in Table 6. Only the reflections with I calc > 2 are given, if not observed. Intensities were estimated visually: vs = very strong; w = weak; vw = very weak. The eight strongest reflections are given in bold.
Single-crystal X-ray diffraction data were collected using a Bruker Smart Breeze diffractometer equipped with an air-cooled Photon II CCD detector, with graphite-monochromatised MoKα radiation (Dipartimento di Scienze della Terra, Università di Pisa). The detector-to-crystal distance was set at 50 mm. A total of 653 frames were collected in ω and φ scan modes in 0.5° slices, with an exposure time of 25 s per frame. Intensity data were corrected for Lorentz-polarisation factors and absorption using Apex3 (Bruker AXS Inc., 2016). Bianchiniite is tetragonal, with unit-cell parameters a = 8.7266(4), c = 15.6777(7) Å and V = 1193.91(12) Å3. The c:a ratio is 1.7965. Reconstructed precession images showed very weak reflections, occurring at low 2θ values and apparently doubling the a parameter (Fig. 3), supporting a superstructure with a’ = 2a and c’ = c. Owing to their weakness, they were ignored and the crystal structure was solved and refined in the tetragonal cell reported above. However, their possible meaning is discussed below.

Fig. 3. Reconstructed precession image of the hk0 reciprocal lattice plane. Weak not-indexed reflections at lower angles can be observed (some of them are indicated by thin dotted arrows). Only positive h and k indices are shown.
The statistical tests on the distribution of |E| values (|E 2 – 1| = 0.877) suggested the occurrence of an inversion center. The examination of systematic absences indicated the space group symmetry I4/mcm. The crystal structure of bianchiniite was solved using Shelxs-97 (Sheldrick, Reference Sheldrick1997) and refined using Shelxl-2018 (Sheldrick, Reference Sheldrick2015). Scattering curves for neutral atoms were taken from the International Tables for Crystallography (Wilson, Reference Wilson1992). During the first step of the crystal structure solution, the positions of Ba, Ti, As, and one anion position were found. Difference-Fourier synthesis maps located three additional anion positions. The isotropic structural model refined to R 1 = 0.038, thus suggesting the correctness of the structural model. Taking into account the electron microprobe data, the site occupancy factors (s.o.f.) at the Ba(1) and As(3) sites were refined using the Ba and As scattering curves only, respectively. The M(2) site is occupied by Ti, V and Fe. Owing to the similar atomic number of Ti (Z = 22) and V (Z = 23) and the slightly higher atomic number of Fe (Z = 26), the s.o.f. was refined using the Ti vs. Fe scattering curves. In addition, taking into account the different crystal-chemical behaviour of Ti and (Fe,V) (with the former giving rise to the off-centre displacement phenomenon; e.g. Megaw, Reference Megaw1968), their positions were refined independently, resulting in the splitting of the M(2) site into M(2a) and M(2b). Titanium was located at M(2a); indeed this position shows the largest polyhedron distortion value, estimated as Δd = (d max – d min), where d max and d min represent the maximum and minimum bond distance, respectively, whereas M(2b) has a more regular coordination environment (see below). A bond-valence calculation performed on the isotropic model indicated the likely mixed (O,F) nature of the O(1) and O(4) sites. Owing to the very similar scattering factors of O (Z = 8) and F (Z = 9), the s.o.f. was fixed on the basis of chemical data and bond-valence requirements. The refinement statistical factor was slightly improved to R 1 = 0.026. Successively, an anisotropic model for cations lowered it to R 1 = 0.016. After several cycles of anisotropic refinement for all atoms, the final statistical factor R 1 converged to 0.0134 for 555 unique reflections with F o > 4σ(F o) and 34 refined parameters. Details of the intensity data collection and crystal structure refinement are given in Table 5. Atom coordinates, s.o.f. and displacement parameters are reported in Table 6, whereas selected bond distances are listed in Table 7. Weighted bond-valence sums, calculated using the bond parameters of Gagné and Hawthorne (Reference Gagné and Hawthorne2015) for cation–oxygen pairs and of Brese and O'Keeffe (Reference Brese and O'Keeffe1991) for cation–fluorine pairs, are shown in Table 8. The crystallographic information file, including reflection data, has been deposited with the Principal Editor of Mineralogical Magazine and is available as Supplementary material (see below).
Table 5. Crystal data and details of data collection and crystal structure refinement for bianchiniite.
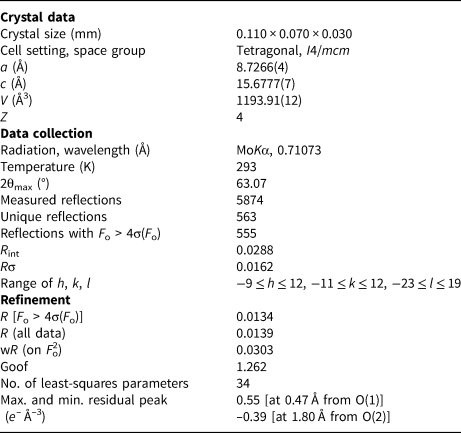
Table 6. Sites, Wyckoff positions, site occupation factors (s.o.f.), fractional atomic coordinates and equivalent isotropic displacement parameters (Å2) for bianchiniite.*

* Note: the refined site scattering at M(2a) + M(2b) matches the site population (Ti0.57V0.22Fe0.21) obtained from electron microprobe analysis.
Table 7. Selected bond distances (in Å) for bianchiniite.

Table 8. Weighted bond-valence balance (in vu) for bianchiniite.*
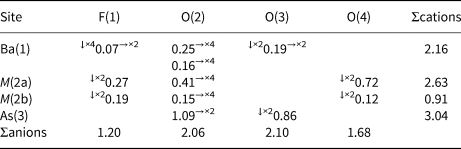
* Notes: left and right superscripts indicate the number of equivalent bonds involving anions and cations, respectively. For sites with mixed site occupancy, the bond valence sums have been weighted.
Crystal structure description
The crystal structure of bianchiniite is formed by columns of corner-sharing M(2) octahedra running along c, connected, in the {001} plane, by (As2O5) dimers. Trigonal pyramidally coordinated As3+ ions form {001} layers, with their apex pointing alternatively up and down. Lone-electron-pair micelles of As3+ atoms alternate, along c, with layers of intercalated Ba atoms (Fig. 4). Consequently, the crystal structure may be described as layered, with heteropolyhedral layers hosting Ba, (Ti,V,Fe) and As atoms, separated by layers occupied by lone-pair electrons and the anion hosted at O(4) connecting successive M(2) octahedra along c.
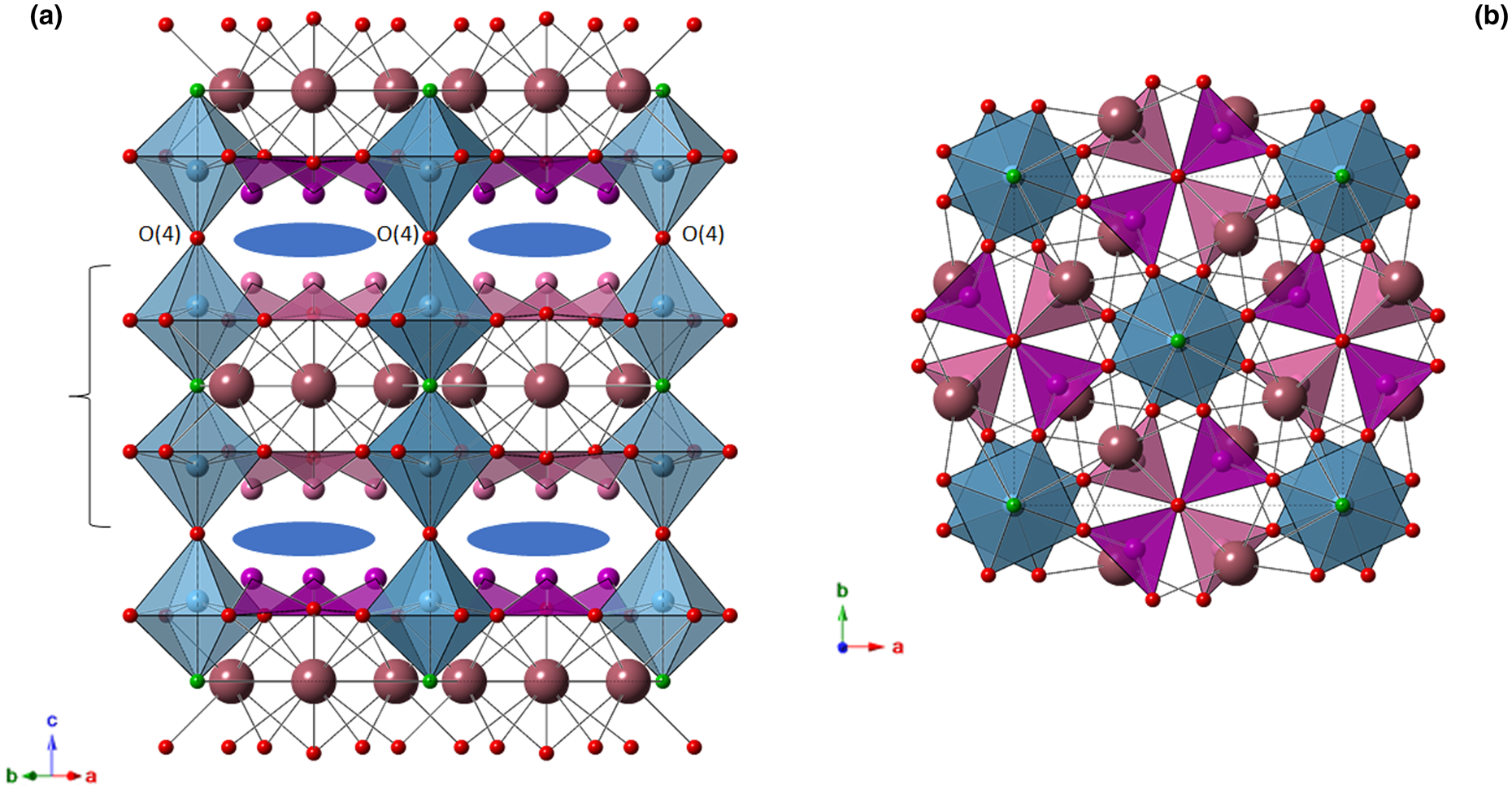
Fig. 4. Crystal structure of bianchiniite as seen down [110] (a) and [001] (b). Symbols: large violet spheres = Ba(1) site; green spheres = F(1) site; red spheres = O(2)–O(4) sites. Light blue octahedra = M(2) site [only the M(2a) position is shown]. For the sake of clarity, As(3) sites belonging to different heteropolyhedral layers are shown in dark magenta and pink, respectively. Blue ellipses indicate the location of As lone-pair electrons. Black dashed lines indicate the unit cell. Bracket indicates one heteropolyhedral layer.
Barium is twelve-fold coordinated, with ten bond distances in the range between 2.79 and 2.98 Å. Two additional bonds with the mixed (F,O) site F(1) at 3.22 Å complete the coordination environment, that can be described as a bicapped pentagonal prism. The average bond distance is 2.942 Å, to be compared with a calculated value of 2.985 Å based on ionic radii for [XII]Ba2+, [IV]O2–, [II]O2– and [II]F– given by Shannon (Reference Shannon1976). The bond-valence sum at the Ba atom is 2.16 vu, in keeping with the expected value.
Arsenic displays the trigonal pyramidal coordination typical of As3+. The <As–O> bond distance, 1.772 Å, agrees with the average distance reported by Majzlan et al. (Reference Majzlan, Drahota, Filippi, Bowell, Alpers, Jamieson, Nordstrom and Majzlan2014), i.e. 1.782 Å. It is worth noting that in the (As2O5) dimers, As–Obr is definitely longer than the two other As–Onbr bonds, i.e. 1.838 and 1.740 Å, respectively. This feature was observed by Cooper and Hawthorne (Reference Cooper and Hawthorne2016) in schneiderhöhnite, where a bridging O atom has a comparatively large valence contribution from non-As atoms. These authors examined the observed contribution in other polymerised As3+ oxysalts (see Table 5 of Cooper and Hawthorne, Reference Cooper and Hawthorne2016), reporting that schneiderhöhnite showed the largest bond-valence contribution from non-As atoms in known minerals (i.e. 0.28 vu). Bianchiniite shows a still larger contribution, represented by the incident bond valence from Ba2+ (0.38 vu), partially compensated through the elongation of the As–Obr distance. The bond-valence sum at the As(3) site is 3.04 vu.
Titanium, along with minor V and Fe (both assumed in the +3 oxidation state – see below), are hosted at the M(2) site. As revealed by the crystal structure refinement, this site is actually split into two sub-positions, namely M(2a) and M(2b). The former is more distorted (Δd = 0.327 Å) than the latter (Δd = 0.140 Å) and this geometrical feature suggests that, as discussed above, Ti4+ may prefer the M(2a) position. The refined site scattering value at M(2a) + M(2b) is 23.04 electrons, to be compared with 23.06 electrons calculated from chemical data. Bond-valence sums at the two split sub-sites was calculated assuming Ti4+ ordered at M(2a) along with minor V3+ and Fe3+, and a mixed (V,Fe) occupancy for M(2b). As it is very difficult to establish a partitioning of V3+ and Fe3+ over these two sub-positions (both elements have a very similar ionic radius, i.e. 0.640 and 0.645 Å, respectively; Shannon, Reference Shannon1976), an equal distribution of both cations at the two split sites was assumed. As a result, the following site populations are proposed (with rounding errors), which match the observed site scatterings: M (2a)(Ti0.57V3+0.08Fe3+0.07) and M (2b)(V3+0.14Fe3+0.14). On this basis, taking into account the ionic radii given by Shannon (Reference Shannon1976), one may expect that M(2a) should have the smaller average bond distance. On the contrary, M(2b) is slightly smaller that M(2a), i.e. 1.969 vs. 1.989 Å, respectively. This discrepancy may be related to the larger degree of polyhedral distortion of M(2a) with respect to M(2b) (the mean bond distance increases with increasing polyhedral distortion; e.g. Bosi, Reference Bosi2014) and the average nature of the ligands coordinating these two split positions. The bond-valence sums at these two split positions [i.e. 2.63 and 0.91 vu, respectively, for M(2a) and M(2b)], agrees with a mixed [Ti4+,(Fe,V)3+] occupancy.
The four independent anion sites have bond-valence sums ranging between 1.20 and 2.10 vu. The O(2) and O(3) sites, with bond-valence sums of 2.06 and 2.10 vu, host O2– anions, whereas the other two sites are underbonded. This underbonding is slight at the O(4) site (1.68 vu), suggesting a partial replacement of O2– by a monovalent anion, whereas at the fourth anion site the underbonding is severe, with 1.20 vu. This supports the dominance of a monovalent anion at this position. These results are in keeping with the occurrence of F, as detected during electron microprobe analysis; accordingly, this anion position was labelled as F(1). The site population at O(4) was fixed at (O0.80F0.20), whereas F(1) was modelled as (F0.70O0.30). According to the s.o.f., it appears that the occupancy of M(2a) is probably related with the occurrence of F– at F(1) and O2– at O(4), whereas when M(2b) is occupied it is associated with O2– at F(1) and F– at O(4).
Discussion
Crystal chemistry of bianchiniite
The proposed end-member formula of bianchiniite corresponds to Ba2(Ti4+V3+)(As2O5)2OF. Barium shows a very minor substitution by Sr (up to 0.04 atoms per formula unit, apfu) and Pb (up to 0.02 apfu), whereas As can be replaced by minor Sb (up to 0.02 apfu). More significant is the replacement of Ti, V and Fe, occurring at the mixed and split M(2) site. The relations between Ti and (V + Fe) content are in Fig. 5a, showing a clear negative correlation that supports the replacement of Ti by (V + Fe).
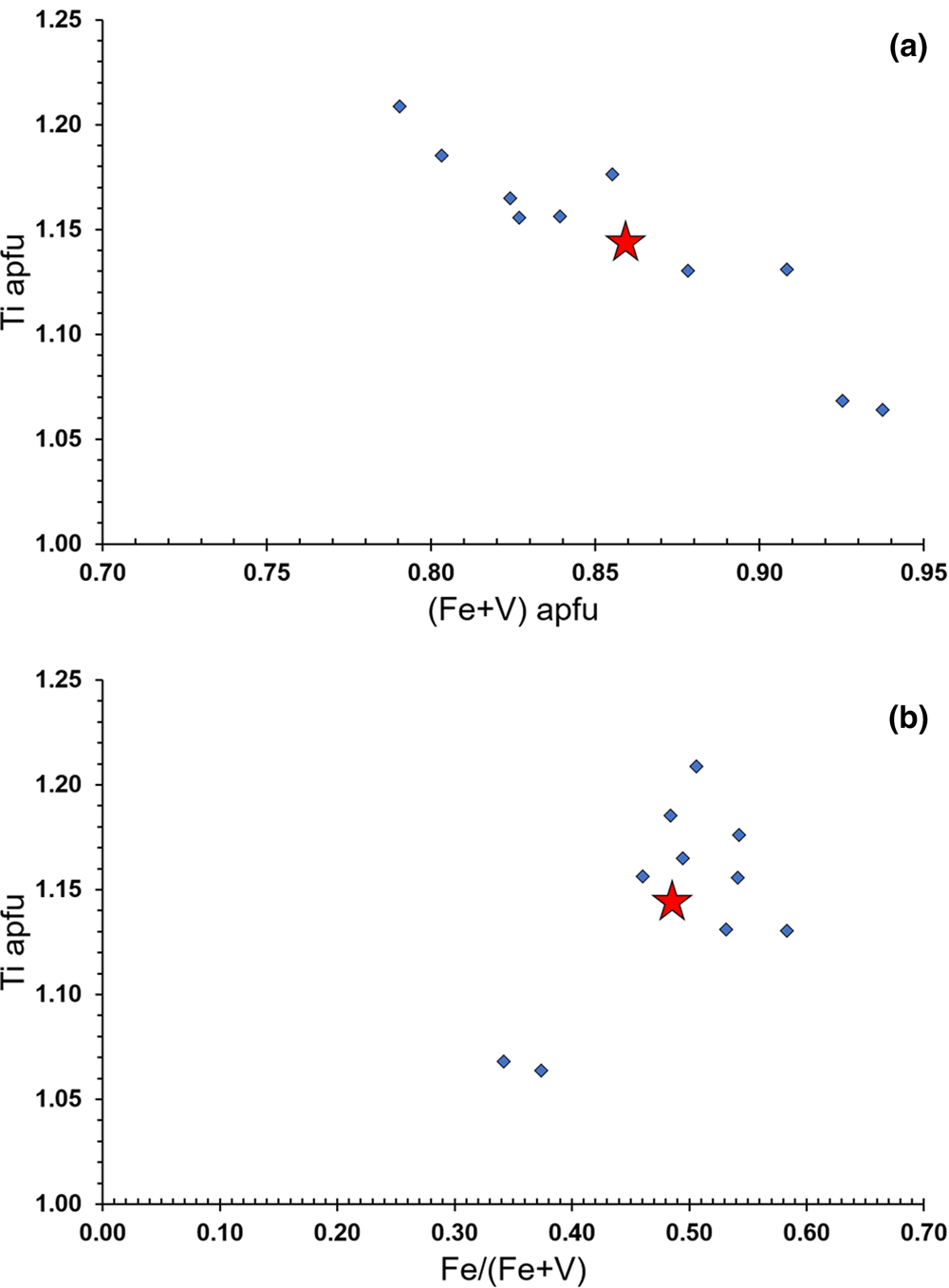
Fig. 5. Relation between Ti and (Fe + V) in apfu (a) and between Ti (in apfu) and the Fe/(Fe + V) atomic ratio (b). Red star indicates the average composition of bianchiniite.
The oxidation state of Fe and V was assumed as +3, on the basis of bond-valence calculations that match the replacement of Ti4+ by trivalent elements. In addition, the F content (0.82 apfu) is consistent with the sum of (V + Fe) (0.86 apfu), thus suggesting the substitution mechanism M (2)Ti4+ + O(1),O(4)O2– = M (2)(Fe,V)3+ + O(1),O(4)F–. However, as V3+ and Fe3+ occur in equal amounts, similar results could be achieved through the occurrence of V4+ and Fe2+. According to Takeno (Reference Takeno2005), the equilibrium between V4+ and V3+ would be in the stability field of Fe2+. Consequently, the M(2) site population (Ti4+0.57V3+0.22Fe3+0.21) could be recasted into (Ti4+0.57V4+0.21V3+0.01Fe2+0.21). Following Bosi et al. (Reference Bosi, Hatert, Hålenius, Pasero, Miyawaki and Mills2019), this would correspond to the end-member formula Ba2(Ti4+1.5Fe2+0.5)(As2O5)2OF, different from the approved one, Ba2(Ti4+V3+)(As2O5)2OF. In addition, the possible average nature of the refined crystal structure would suggest the occurrence of two different M(2) positions, one preferentially occupied by Ti4+, the other by a mixed (V,Fe) population. In such a case, the formula of bianchiniite could also be written as Ba2Ti(V4+0.5Fe2+0.5)(As2O5)2OF. Unfortunately, these distinct site populations are virtually indistinguishable using available data; for instance, the calculated <M(2)–Φ> (Φ = O2–, F–) distance and bond-valence sums for both cation distributions are very similar. Only spectroscopic techniques (e.g. Mössbauer and Vis-NIR spectroscopies) could solve this issue, if more material will become available. For this reason, the IMA-CNMNC approved formula is retained, assuming the coexistence of V3+ and Fe3+.
The average content of V and Fe supports a very subtle dominance of the former over the latter (Table 3). The examination of the spot analyses showed Fe/(V+Fe) atomic ratios ranging between 0.34 and 0.58, with an average value of 0.48(8) (Fig. 5b). This means that both V and Fe can be dominant constituents in bianchiniite. In the definition of the new mineral species, V was assumed as the species-forming constituent as it has the wider chemical range with respect to the 50% boundary. Consequently, taking into account the current chemical range of bianchiniite, the use of only one name also applies to chemical domains having a slight Fe-dominance, in agreement with the IMA-CNMNC rules (Nickel, Reference Nickel1992; Hatert and Burke, Reference Hatert and Burke2008). However, additional findings could allow the description of the Fe-dominant end-member of bianchiniite or a revision of its formula based on spectroscopic data.
Relations with other species
The crystal structure of bianchiniite is a novelty among diarsenite minerals. Moreover, it is the first naturally occurring Ba-arsenite, although minor Ba occurs in graeserite (Biagioni et al., Reference Biagioni, Bonaccorsi, Perchiazzi, Hålenius and Zaccarini2020a).
Bianchiniite has some resemblance with fresnoite (Masse et al., Reference Masse, Grenier and Durif1967; Moore and Louisnathan, Reference Moore and Louisnathan1969; Markgraf et al., Reference Markgraf, Halliyal, Bhalla, Newnham and Prewitt1985; Krzątala et al., Reference Krzątala, Krüger, Galuskina, Vapnik and Galuskin2021) and with melilite-group minerals (Bindi et al., Reference Bindi, Nespolo, Krivovichev, Chapuis and Biagioni2020). Fresnoite, ideally Ba2Ti(Si2O7)O, has unit-cell parameters a = 8.52 and c = 5.21 Å, with space group P4bm. Its crystal structure shows ten-fold coordinated Ba atoms and five-fold coordinated Ti, along with disilicate groups (Fig. 6). As first noted by Moore and Louisnathan (Reference Moore and Louisnathan1969), this mineral displays some structural features in common with melilite-group phases. Among them, the recently described mineral bennesherite, Ba2Fe2+Si2O7, is the first Ba member of this group (Krzątala et al., Reference Krzątala, Krüger, Galuskina, Vapnik and Galuskin2021). Its unit-cell parameters are a = 8.23 and c = 5.29 Å with space group P ${\bar 4}$ 21m. In agreement with the general formula of melilite-group minerals, X 2T(1)[T(2)2O7] (e.g. Bindi et al., Reference Bindi, Nespolo, Krivovichev, Chapuis and Biagioni2020), the T(1) tetrahedron is occupied by Fe2+, whereas Si4+ is hosted at T(2) (Fig. 6). In bianchiniite, the T(1) tetrahedron of melilite-group minerals and the five-fold coordinated Ti-centred site of fresnoite are replaced by the mixed and split (Ti,V,Fe)-centred M(2) octahedron, forming columns along c. Disilicate groups (Si2O7)6– occurring in fresnoite- and melilite-group minerals are replaced by diarsenite groups (As2O5)4–.
21m. In agreement with the general formula of melilite-group minerals, X 2T(1)[T(2)2O7] (e.g. Bindi et al., Reference Bindi, Nespolo, Krivovichev, Chapuis and Biagioni2020), the T(1) tetrahedron is occupied by Fe2+, whereas Si4+ is hosted at T(2) (Fig. 6). In bianchiniite, the T(1) tetrahedron of melilite-group minerals and the five-fold coordinated Ti-centred site of fresnoite are replaced by the mixed and split (Ti,V,Fe)-centred M(2) octahedron, forming columns along c. Disilicate groups (Si2O7)6– occurring in fresnoite- and melilite-group minerals are replaced by diarsenite groups (As2O5)4–.

Fig. 6. Projections of the crystal structures of bianchiniite along [001] (a) and [110] (b), fresnoite (c, d) and a melilite-group mineral (e, f). Only one heteropolyhedral layer of bianchiniite is shown. Same symbols as in Fig. 4. Silicon-centred tetrahedra are shown in blue, whereas Ti and T(1) sites in fresnoite- and melilite-group minerals are light blue. The Ba and X cation are shown as violet and light blue spheres, respectively. In (g), the crystal structure of synthetic Ba3Ti2Si4O14Cl is shown as seen along [110]. The two symmetry-independent Si-centred sites are shown as light and dark blue tetrahedra. Titanium-centred octahedra and Ba atoms are shown as in bianchiniite. O and Cl are shown as green and red spheres, respectively.
The c unit-cell parameters of fresnoite- and melilite-group minerals are very similar, usually ~5 Å (see table 1 in Krzątala et al., Reference Krzątala, Krüger, Galuskina, Vapnik and Galuskin2021); such a periodicity is related to the alternation, along c, of (Ti/Si) and [T(1)/T(2)] layers with intercalated Ba and X layers in fresnoite- and melilite-group minerals, respectively. In bianchiniite, the c parameter is tripled. Indeed, whereas in fresnoite- and melilite-group minerals every cell contains one layer of Ba and X cations and one layer of T(1) and T(2) sites, in the unit-cell of bianchiniite two heteropolyhedral layers occur, separated by two lone-electron-pair micelles. The layers are staggered, owing to a rotation of 45° of successive heteropolyhedral layers stacked along c.
The heteropolyhedral layer occurring in bianchiniite (Fig. 4) is topologically similar to one of the two constitutive layers of the synthetic compound Ba3Ti2Si4O14Cl (Abeysinghe et al., Reference Abeysinghe, Smith and zur Loye2017). This phase is tetragonal, space group P4/mbm. Its a unit-cell parameter is similar to that of bianchiniite, i.e. 8.67 Å, whereas the c axis is longer, i.e. 18.65 Å. In the synthetic compound, the bianchiniite-like layer is formed by split TiO5Cl octahedra, Si2O7 groups, and intercalated twelve-coordinated Ba atoms. The O/Cl disorder related to the occupancy of the split Ti sub-sites is similar to the O/F disorder in bianchiniite. The bianchiniite-like layer alternates with another kind of layer formed by TiO5 square pyramids and by Si4O12 rings, along with eleven-fold coordinated Ba atoms (Abeysinghe et al., Reference Abeysinghe, Smith and zur Loye2017). In agreement with Makovicky (Reference Makovicky1997), this may suggest the possible existence of merotypic relations between bianchiniite and synthetic Ba3Ti2Si4O14Cl.
As shown in Fig. 3, some very weak reflections indicate the presence of a superstructure. The occurrence of incommensurate superstructure reflections is a common feature of melilite-group minerals (e.g. Bindi et al., Reference Bindi, Nespolo, Krivovichev, Chapuis and Biagioni2020). In some cases, a commensurate superstructure has been proposed. This is the case of another mineral probably related to melilite-group phases, jeffreyite, (Ca,Na)2(Be,Al)Si2(O,OH)7 (Grice and Robinson, Reference Grice and Robinson1984). This phase has a pseudotetragonal subcell, with a = b = 7.5 Å and c = 5.0 Å; the real symmetry seems to be orthorhombic, C2221, with a = 14.90, b = 14.90 and c = 40.41 Å. In this case, the c periodicity is eight-times the basic 5 Å periodicity. In addition, a superstructure along a and b has been observed. This feature parallels the possible 2a superstructure observed in precession images of bianchiniite (Fig. 3). Grice and Robinson (Reference Grice and Robinson1984) hypothetically attributed superstructure reflections to some ordering of Na, Al and (OH) groups in the still unsolved crystal structure of jeffreyite. In bianchiniite, the ordering of Ti, V and Fe at the M(2) site, suggested also by the split nature of this position, may be invoked to explain the appearance of such superstructure reflections.
Conclusion
For the last ten years, the Monte Arsiccio mine has enriched the mineral systematics with several new mineral species belonging to different crystal-chemical classes, i.e. sulfosalts, oxides and sulfates. Bianchiniite is probably one of the most remarkable discoveries, due to its unique chemistry, mirroring some geochemical features characterising the pyrite ± baryte ± iron-oxide ore deposits of the Apuan Alps. In addition to baryte, a few other minerals having Ba as a primary constituent are currently known: benstonite, cymrite and mannardite – all identified from the Monte Arsiccio mine (Biagioni et al., Reference Biagioni, Orlandi and Pasero2009; Biagioni and Orlandi, Reference Biagioni and Orlandi2010). Similarly to mannardite, Ba in bianchiniite is associated with Ti and V, with the latter element being another geochemical fingerprint for this kind of ore deposit from the Apuan Alps, occurring in mineral species at both the Monte Arsiccio and the Buca della Vena mines (e.g. Merlino and Orlandi, Reference Merlino and Orlandi1983; Mellini et al., Reference Mellini, Orlandi and Vezzalini1986; Biagioni et al., Reference Biagioni, Bonaccorsi, Perchiazzi, Hålenius and Zaccarini2020a). The crystallisation environment of bianchiniite was also enriched in As, in keeping with the As-rich nature of the Monte Arsiccio ore deposit.
In addition to the geochemical implications, the finding of bianchiniite reveals the flexibility of melilite-like structures, with diarsenite groups replacing disilicate groups and with the tetrahedrally-coordinated T(1) cations substituted by octahedrally-coordinated cations, in a novel and unpredicted crystal structure arrangement for natural compounds.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2021.28
Acknowledgements
The paper benefited of the careful revision of Joel D. Grice, Luca Bindi, Yves Moëlo and one anonymous reviewer. Mario Bianchini and Stefano Magnanelli are thanked for providing us with the specimen of bianchiniite. Luca Bindi is acknowledged for useful discussion on melilite-group minerals. This research was financially supported by the Ministero dell'Istruzione, Università e Ricerca through the project PRIN 2017 “TEOREM – deciphering geological processes using Terrestrial and Extraterrestrial ORE Minerals”, prot. 2017AK8C32.

















 Open access
Open access