


The epigenetic mechanism of DNA methylation controls gene expression and repression(Reference Lim and Maher1). DNA methylation refers to the covalent addition of methyl (CH3) groups to the C5 position of the pyrimidine ring of cytosines, typically in a CpG dinucleotide, of which there are approximately 28 million in the haploid genome of a human subject(Reference Stevens, Cheng and Li2). Regions of DNA with a high CpG content are referred to as CpG islands (CGI). There are approximately 45 000 CGI per human subject haploid genome(Reference Antequera and Bird3), which are typically between 200 and 1400 bp in length(Reference Larsen, Gundersen and Lopez4) and generally located around transcription start sites(Reference Saxonov, Berg and Brutlag5). Saxonov et al. determined that 72 % of promoters are rich in predominantly unmethylated CpG(Reference Saxonov, Berg and Brutlag5). DNA methylation varies due to a number of factors including age and disease status. Interestingly, hypermethylation of CpG sites in promoters or enhancers typically leads to transcriptional silencing, whereas hypomethylation of CpG sites in a gene body frequently results in an increase in gene expression(Reference Yang, Han and De Carvalho6, Reference Mendizabal and Yi7).
As outlined in Fig. 1, the production of 5-methylcytosine is regulated by DNA methyltransferases (DNMT) DNMT1, DNMT3a and DNMT3b, which transfer CH3 groups from S-adenosyl-l-methionine. DNMT1 primarily acts as a maintenance methyltransferase, targeting hemimethylated DNA, formed after DNA replication, thus ensuring the re-establishment of the parental DNA methylation pattern in daughter DNA(Reference Goyal, Reinhardt and Jeltsch8); while DNMT3a and DNMT3b act as de novo DNMT(Reference Okano, Bell and Haber9). Additionally, there is another member of the DNMT3 family, DNMT3L. Although catalytically inactive, DNMT3L has been observed to markedly stimulate the de novo methylation of DNA by DNMT3a when coexpressed(Reference Chédin, Lieber and Hsieh10).
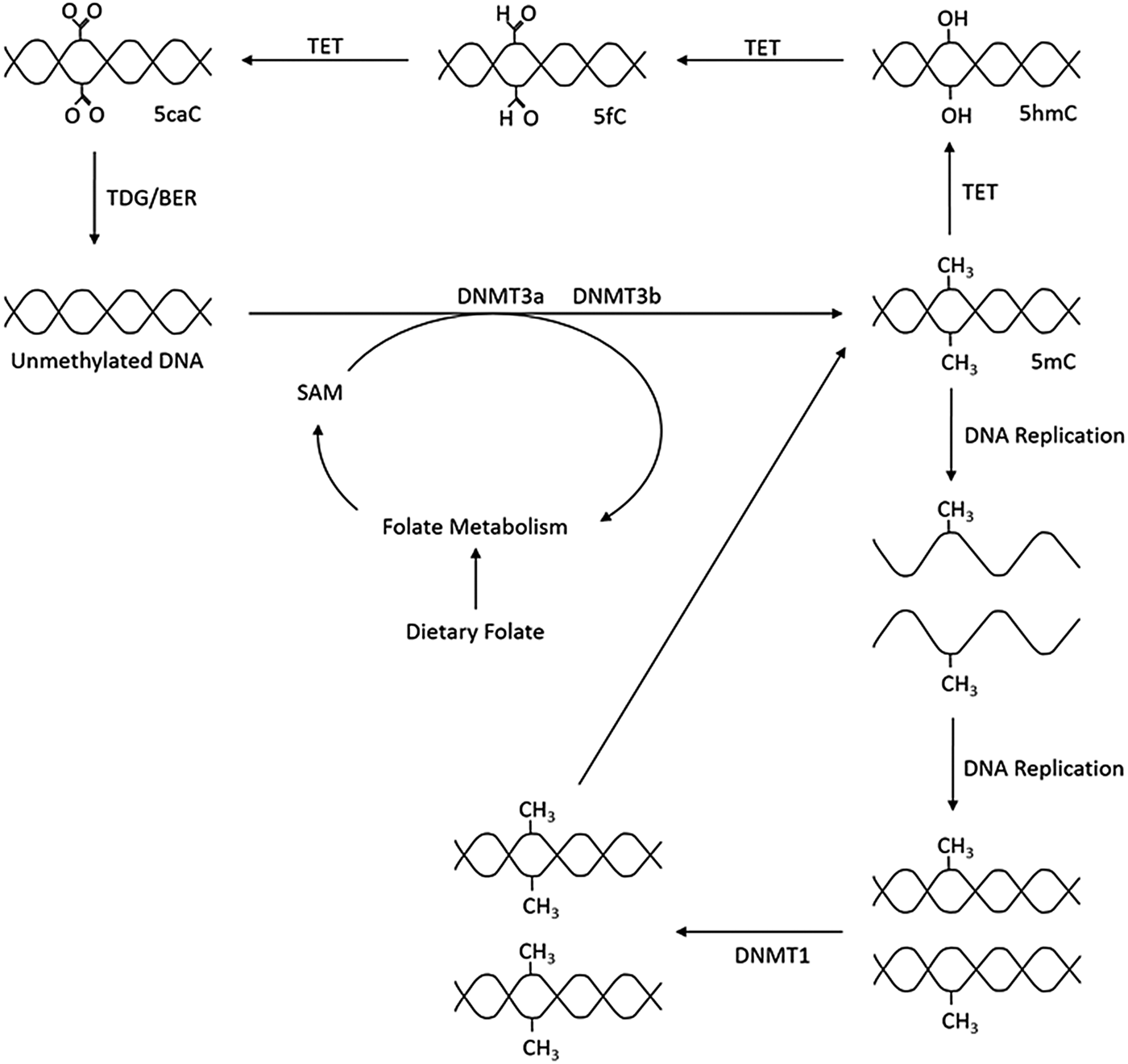
Fig. 1. Overview of DNA methylation. De novo methylation is regulated by DNA methyltransferase (DNMT)3a and DNMT3b and uses S-adenosyl methionine (SAM) as a methyl donor. The new methylation pattern is passed on to daughter cells through DNMT1, which acts on hemi-methylated DNA. DNA can become demethylated through the ten–eleven translocation (TET) and thymine DNA glycosylase (TDG) enzymes and base excision repair (BER). 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethlcytosine; 5fC, 5-formylcytosine; 5caC, 5-carboxylcytosine.
Demethylation can be either passive, through incorrect DNA replication, or be an active process regulated by ten–eleven translocation (TET) enzymes. TET catalyse the oxidation of 5-methylcytosine to 5-carboxylcytosine via the intermediates 5-hydroxymethlcytosine and 5-formylcytosine. Thymine DNA glycosylase then removes 5-carboxylcytosine and 5-formylcytosine from the DNA strand allowing the insertion of an unmethylated cytosine into the deleted base site through base excision repair(Reference Rasmussen and Helin11).
Impact of ageing on DNA methylation
During ageing, epigenetic drift can be used to describe the increase in methylation in CGI sites which are unmethylated in the young, and the decrease in methylation globally. These findings have been reported across species(Reference Maegawa, Lu and Tahara12). Maegawa et al. showed that average methylation increased from 2 (sem 0·1) to 18 (sem 5), 2 (sem 0·3) to 22 (sem 3) and 3 (sem 0·5) to 20 (sem 4) % with age in sites observed to be unmethylated in young mice, rhesus monkeys and human subjects, respectively. When analysing highly methylated non-CGI sites, ageing resulted in a reduction in methylation from 94 (sem 0·4) to 78 (sem 4), 94 (sem 0·3) to 73 (sem 4) and 93 (sem 1) to 74 (sem 2) % in the same three mammalian species. These data indicate that methylation drift associated with ageing is evolutionarily conserved. Interestingly, drift rates were calculated as 4·1 (sem 1·2), 0·34 (sem 0·14) and 0·1 (sem 0·02) % per year for mice, rhesus monkeys and human subjects, respectively, and an inverse relationship between the rate of methylation drift and longevity in these mammalian species was established(Reference Maegawa, Lu and Tahara12). A similar finding was described by Wilson et al.(Reference Wilson, Smith and Ma13). In this work, ageing resulted in a global decrease in 5-methyldeoxycytidine in multiple tissues from two murine models and human bronchial epithelial cells obtained from autopsy donors, and an inverse relationship between lifespan and rate of loss of 5-methyldeoxycytidine was reported. An estimated loss of 5·6–8·9 × 105 and 2·3–2·8 × 105 per year was observed for Mus musculus and Peromyscus leucopus species, which have lifespans of 3·5 and 8·0 years, respectively, while a loss of 1·6 × 104 per year was observed in human cells. Their conclusion corroborates with the findings of Drinkwater et al. (Reference Drinkwater, Blake and Morley14) where it was determined that lymphocytes obtained from 20–30-year-old volunteer donors contained 54·6 (sem 1·6) % methylated CmCGG sites, while a statistically significant 7·1 % reduction (47·5 (sem 2·6) %) was observed in 65–80 year olds.
Maegawa et al. further examined if methylation drift is ubiquitous in differing tissue types. By analysing twelve genes that were associated with hypermethylation and three associated with hypomethylation with age, it was determined that tissue from kidney and liver generally exhibited lower levels of age-related hypermethylation. In contrast, tissue from the intestines (small and large) and bone marrow showed reduced age-associated hypomethylation. In further investigations by Maegawa et al., it was reported that there was a significant inverse relationship between methylation drift and the level of change in gene expression. It was noted that when looking at the methylation pattern of genes that had increased expression, a significant reduction in DNA methylation was observed, while conversely, silenced genes had a concomitant increase in methylation(Reference Maegawa, Lu and Tahara12).
Altered expression of the enzymes responsible for DNA methylation and demethylation have been repeatedly reported to be a contributing factor to changes observed in DNA methylation patterns with age. For instance, Sun et al. observed the expression of genes encoding for the DNMT DNMT1, DNMT3a and DNMT3b, considerably declined between the ages of 4 and 24 months in C57BL/6 male mice. Interestingly, the expression of demethylation enzymes tet1 and tet3 was also reduced with age(Reference Sun, Luo and Jeong15). In human subjects, a reduction in TET1 and TET3 expression was observed with age and a correlation between TET1 and DNMT1, DNMT3b and thymine DNA glycosylase was determined in peripheral blood mononuclear cells obtained from 188 volunteers, aged 34–74 years, from eight European countries. Interestingly, while a global reduction in 5-hydroxymethylcytosine was also detected with age, a statistically significant increase in the methylation of the CGI within the TET1 gene was found in 69–74 year olds when compared with 34–41 year olds(Reference Valentini, Zampieri and Malavolta16). These findings are consistent with the observation that hypermethylation within certain gene regions is often associated with gene silencing.
In contrast to the findings of Sun et al., Lopatina et al. showed that although DNMT1 declined with age in WI-38 fibroblast cells, the activity of de novo methylation enzymes decreased in middle age, compared with young cells, and rose slightly with senescence. This resulted in the ratio of de novo to maintenance methylation enzymes increasing with age. The authors postulate that the decline in DNMT1 could lead to the global hypomethylation, and add that the rise in the ratio of de novo to maintenance methylation enzymes with age could be responsible for the regional hypermethylation associated with gene silencing(Reference Lopatina, Haskell and Andrews17). Similarly, Casillas et al. observed that DNMT1 expression declined significantly with age in fetal human subjects WI-38 fibroblasts, with old aged cells having 75·44 % of the expression exhibited by young cells. Furthermore, the activity of this maintenance methyltransferase declined from 83·2 cpm/μg protein in young cells to 52·1 cpm/μg protein in middle-aged cells, and to 28·1 cpm/μg protein in old lung WI-38 fibroblast cells. Conversely, the activity of the de novo methyltransferases increased from 21·4 cpm/μg protein in young cells to 59·0 and 75·0 cpm/μg protein in middle-aged and old cells, respectively. Interestingly, ageing appeared to have an opposing effect on the expression of the de novo methyltransferases, with DNMT3a declining to 60·61 % that of young cells in old age, while the expression of DNMT3b in young cells was 75·21 % compared with that expressed in old cells. Thus, a change in the ratio between maintenance methyltransferases and de novo methyltransferases could be a key factor in the aberrant DNA methylation associated with ageing.
DNA methylation and cancer
Germline cells have specific DNA patterns to enable suitable gene regulation during embryonic development. Importantly, within a small proportion of genes, one parental allele is exclusively expressed, due to DNA methylation regulated gene imprinting(Reference Barlow and Bartolomei18). Inappropriate methylation during development can result in imprinting failures and diseases including Beckwith–Wiedemann, Prader–Willi, Silver–Russell and Angelmans’ syndromes(Reference Bartolomei and Ferguson-Smith19). Epigenetic modifications are also frequently seen in diseases with later onsets including cancer(Reference Kulis and Esteller20), neurodegeneration(Reference Sanchez-Mut, Heyn and Vidal21) and autoimmune disease(Reference Richardson22). With a focus on the effect of the epigenetic modification on cancer pathogenesis, both gene silencing, due to hypermethylation in gene promoters(Reference Merlo, Herman and Mao23), and oncogene activation or chromosomal instability due to global hypomethylation(Reference Gaudet, Hodgson and Eden24, Reference Rosty, Ueki and Argani25) will be discussed.
In one study of promoter hypermethylation, it was reported that in seven of nine non-small cell lung cancers, the tumour suppressor gene p16 was fully methylated, while in the CGI in the samples of healthy lung, kidney and blood lymphocytes, the CpG sites were found to be unmethylated(Reference Merlo, Herman and Mao23). Interestingly, Christensen et al. discovered through locus-by-locus analysis, a trend between loci methylation and cancer characteristics, including tumour grade and size, oestrogen and progesterone status and triple negative status in invasive breast cancer specimens, from 162 women from Northern California. Interestingly, at all seventy-four CpG loci, which were associated with tumour size, there was a positive correlation between the level of methylation and tumour size. Moreover, increased methylation was observed in all five CpG loci associated with lymph node infiltration, when disease-positive lymph nodes were reported. Array validation revealed CpG within the promoters of P2RX7, a gene encoding for a receptor which mediates apoptosis, and HSD17B12, a gene coding for an enzyme involved in oestrogen metabolism and fatty acid elongation, had statistically elevated methylation levels as tumour size increased, while methylation of CpG within the promoter of GSTM2, which reduced mRNA expression of the detoxifying enzyme GSTM2, was correlated with tumour grade(Reference Christensen, Kelsey and Zheng26).
Similarly to the aberrant DNA methylation associated with ageing, disease-associated changes to the methylome could be due to changes in DNMT expression. For instance, in a study of seventy-six women with primary cervical cancer, DNMT1 was on average observed in 77·5 % of cancer cells, in comparison with only 16 % of normal cells. In addition, the intensity score was calculated as 1·0 for cancerous cells compared with a reduced figure of 0·2 for normal cells. Interestingly, individuals with >77·5 % DNMT1-positive cells were 4·3 times more likely to die prematurely compared with individuals who exhibited <77·5 % DNMT1-positive cells, while those with an intensity score >0·9625 were 4·9 times more likely to die earlier than those with an intensity score <0·9625(Reference Piyathilake, Badiga and Borak27). Furthermore, Mizuno et al. determined that in thirty-three patients with acute myeloid leukaemia, DNMT1, DNMT3a and DNMT3b exhibited an average 5·3-, 4·4- and 11·7-fold increase in comparison with the levels observed in control bone marrow cells(Reference Mizuno, Chijiwa and Okamura28). Interestingly, p15INA4B, a tumour suppressor gene commonly silenced by methylation in acute myeloid leukaemia, was methylated in 72 % of acute myeloid leukaemia patients, and in these twenty-four cases, DNMT1 was statistically higher than those without p15INA4B methylation. Further examination of chronic myeloid leukaemia cells revealed that DNMT expression was phase dependent. During the chronic phase, the expression of these three methyltransferases was comparable with normal bone marrow cells; however, with advancement to the acute phase, DNMT1, DNMT3a and DNMT3b expression were raised with an average 3·2-, 4·5- and 3·4-fold increase, respectively(Reference Mizuno, Chijiwa and Okamura29). Conversely, Gaudet et al. reported that mice exhibiting 10 % of DNMT1 compared with wild-type mice exhibited a 30 % reduction in birth weight, and 80 % developed aggressive T-cell lymphoma within 8 months(Reference Gaudet, Hodgson and Eden24). While examining hypomethylated tumours, it was determined that ten of twelve exhibited chromosomal instability (gain of chromosome 15), in comparison with only two of twelve Moloney murine leukaemia virus-induced tumours, thus indicating that global hypomethylation can also play a role in the pathogenesis of cancer through chromosomal instability.
EN1 gene and disease
The EN1 gene encodes for the homeobox protein engrailed-1. First characterised in Drosophila, EN1 mutation results in abnormal development including posterior–anterior duplications and malformation of the wings(Reference Garcia-Bellido and Santamaria30). Within human subjects, the EN1 gene has been associated with pattern formation within the central nervous system during development(Reference Zec, Rowitch and Bitgood31). Wilson et al. detail that the expression of EN1 is observed within multiple neuronal cell types within the cerebellum and that significant changes to its distribution occurs during gestation, with expression remaining until >21 d(Reference Wilson, Kalinovsky and Orvis32).
Hypermethylation of this gene has been observed in multiple cancer types including colorectal(Reference Mayor, Casadomé and Azuara33), prostate(Reference Devaney, Stirzaker and Qu34) and breast cancer(Reference Carrascosa, Sina and Palanisamy35). For instance, Bell et al. reported that the EN1 gene transcriptional start site exhibited significant hypermethylation in human subjects salivary gland adenoid cystic carcinoma when compared with normal tissue, with a 59 % difference in methylation across the EN1 promoter. Furthermore, the extent of hypermethylation was correlated with tumour grading, location and patient outcome. Significantly, it was observed that out of thirty-two loci, the EN1 gene displayed the greatest difference in methylation between normal and diseased tissue, and little variation in hypermethylation across nine CGI, thus emphasising its possible use as a biomarker in cancer(Reference Bell, Bell and Weber36). Similarly for prostate cancer, differential methylation between normal and cancerous cells was greatest in the EN1 gene(Reference Devaney, Stirzaker and Qu34). In addition, the EN1 gene was most frequently methylated in colorectal cancer when compared with the SCTR and INHBB genes. Interestingly, EN1 was more likely to be methylated in colorectal carcinoma compared with colorectal adenoma, with 73 % (66/90) colorectal carcinomas and 40 % (four of ten) adenomas showing hypermethylation, and result in gene silencing(Reference Mayor, Casadomé and Azuara33). Similarly, Frigola et al. determined that the EN1 gene was hypermethylated in 70 % of colorectal tumours, and found that hypermethylation resulted in the suppression of the EN1 gene(Reference Frigola, Song and Stirzaker37). Importantly, Mayor et al. outlined that only 1·12 % (one of eighty-nine) of EN1 genes in normal samples exhibited hypermethylation, an important factor when searching for a cancer biomarker. Interestingly, EN1 methylation resulted in approximately a 30 % reduced survival rate after 5 years compared with patients without hypermethylation of the EN1 gene(Reference Mayor, Casadomé and Azuara33).
Effect of poor diet on DNA methylation and disease
There is a strong association between poor diet, obesity and cancer(Reference Dobbins, Decorby and Choi38). For instance, Zhang et al. examined the effect of DNA methylation in rats fed a high-fat diet for 14 weeks, and reported that within 1000 bp of transcriptional start sites of known genes, seven genes exhibited differentially methylated CpG. Differential CpG methylation ranged from 5 to 22 % difference, in addition to altering gene expression, in animals which gained approximately 90 % body mass from the high-fat diet in comparison with rats fed a standard chow diet. When expanding to CpG within 10 000 bp of transcriptional start sites, 147 genes were differentially methylated and expressed. One of the genes of note, Phlda1, became hypermethylated with a high-fat diet, which was associated with reduced expression, and in turn steatosis, a contributor to the pathophysiology of obesity(Reference Zhang, Chu and Dedousis39). Furthermore, Vucetic et al. outlined that mice fed a high-fat diet (60 % fat) from weaning at 3 weeks, until 18–20 weeks, showed significant hypermethylation in the μ-opioid receptor promoter in reward-related brain regions, and repression of the μ-opioid receptor gene, which was related to an increase in binding of the transcriptional repressor CH3 CpG binding protein 2. It was suggested that repression of the μ-opioid receptor gene was responsible for a significantly reduced preference for sucrose; thus indicating that animals on a high-fat diet exhibit reward hypofunctioning, which may contribute to difficulties reversing obesity after long-term exposure to highly palatable foods(Reference Vucetic, Kimmel and Reyes40). As mentioned, obesity is strongly linked to cancer(Reference Han, Stevens and Truesdale41). For instance, it has been found that many of the thirty-one differentially methylated CpG in obese children, and 151 differentially methylated CpG in severely obese children discussed by Fradin et al. are also associated with cancer, thus warranting concern regarding the risk for cancer pathogenesis in later life(Reference Fradin, Boëlle and Belot42). Similar results were observed by Xu et al., who examined differentially methylated CpG sites in forty-eight obese African American participants aged 14–20 years compared with their non-obese counterparts(Reference Xu, Su and Barnes43). It is important to note that the type of ingested fat may differentially methylate DNA. Garcia-Escobar et al. examined the effect of different fats on TNF-α promoter methylation and reported reduced methylation in animals who were fed coconut oil (high SFA), which was inversely correlated with the pro-inflammatory cytokine TNF-α in adipocytes(Reference García-Escobar, Monastero and García-Serrano44).
Aberrant DNA methylation therapy
There is increasing evidence suggesting the influence of lifestyle factors such as diet, physical activity, weight and smoking status, on the methylome and age-related disease. Due to the ability to somewhat modulate and reverse methylation using lifestyle factors, targeting the methylome more rigorously with chemotherapy could provide a promising avenue to treat diseases such as cancer.
Diet
The role of diet in modulating metabolic health throughout lifespan has long been known. For instance, a significant amount of insight has been gained from analysing the impact of being born during the Dutch Hunger Winter, which took place in the Netherlands during World War II. It is now emerging that the changes to DNA methylation could be a central player in directing how the deleterious effects of the Dutch Hunger Winter unfold. A recent genome-scale analysis of differential DNA methylation in whole blood after periconceptional exposure to famine during the Dutch Hunger Winter emphasises this phenomenon(Reference Tobi, Goeman and Monajemi45). Following a thorough assessment of prenatal malnutrition-associated differentially methylated regions, it was found that prenatal malnutrition-associated differentially methylated regions, which preferentially occur at regulatory regions, are characterised by intermediate levels of DNA methylation, and map to genes enriched for differential expression during early development. Moreover, it was revealed that differential methylation of prenatal malnutrition-associated differentially methylated regions was associated with pathways that are defined by growth and metabolism. Prenatal malnutrition-associated differentially methylated regions found in the insulin receptor precursor gene and the carnitine palmitoyltransferase 1A gene (involved in fat metabolism) were found to have enhancer activity in vitro and differential methylation was interconnected with birth weight and serum low-density lipoprotein-cholesterol levels. In addition to the findings from studying those exposed to the Dutch Hunger Winter, it has also been recognised by Barker since the mid-1990s that exposure to a suboptimal intra-uterine environment has deleterious metabolic consequences for later life(Reference Barker46). Similar to the Dutch Hunger Winter, recent studies have revealed that this phenomenon is underpinned by epigenetic regulation. For instance, it has been shown that placental leptin gene DNA methylation levels were correlated with glucose levels (2 h post-oral glucose tolerance test) in women with impaired glucose tolerance and with decreased leptin gene expression in the whole cohort(Reference Bouchard, Thibault and Guay47). The methylome is not simply a nutrient sensor during the intra-uterine period. Strikingly, in a recent study, DNA methylation changes were correlated with body composition in pre-school children as part of the epigenome-wide-analysis in the European Childhood Obesity Project Study. It was found that DNA methylation variants were identified to be associated with BMI, fat mass, fat-free mass, fat mass index and fat-free mass index(Reference Rzehak, Covic and Saffery48). Specific aspects of diet have also been associated with DNA methylation changes. As discussed, the effect of poor diet on aberrant DNA methylation and disease pathogenesis can be significant; therefore, it is conceivable that a healthy diet could play a role in the prevention of aberrant DNA methylation. For instance, plant polyphenols, originating in fruit and beverages often associated with healthy diets, have been associated with reduced oxidative stress, inflammation and risk of cancer(Reference Zhang and Tsao49), which could be mediated through modulation of DNA methylation(Reference Mileo and Miccadei50). In one example, polyphenols associated with the Mediterranean Annuraca apple, reportedly increased p53, reduced methylation in the promoters of hMLH1, p14ARF and p16INK4a, restoring normal expression of silenced tumour suppressor genes in colorectal cancers(Reference Fini, Selgrad and Fogliano51). In another example, it was observed that 2 weeks of cocoa (6 g/d), a rich source of polyphenols, led to 2·991 (sd 0·366) % 5-methylcytosine in comparison with 3·909 (sd 0·380) % 5-methylcytosine in peripheral leucocyte DNA from control participants with pre-hypertension, type 1 hypertension or hypercholesterolaemia in a randomised controlled trial. Furthermore, in vitro treatment of subject peripheral blood mononuclear cells revealed cocoa significantly lowered DNMT1, 3a and 3b mRNA expression in addition to methylenetetrahydrofolate reductase and 5-methyltetrahydrofolate-homocysteine methyltransferase reductase gene expression(Reference Crescenti, Solà and Valls52). Similar results were observed by Nandakumar et al. who reported that green tea polyphenols epicatechin-gallate and epigallocatechin-3-galate significantly lowered DNMT1, 3a and 3b activity and expression in a dose-dependent manner, reduced global methylation and reactivated the silenced tumour suppressor genes p16INK4a and Cip1/p21 in human subjects skin cancer A431 cells(Reference Nandakumar, Vaid and Katiyar53).
Folate feeding studies
One of the most studied supplements in regards to DNA methylation is folate. This is because, folate plays a key role in C1 metabolism through its conversion to N-5-methyltetrahydrofolate, which in turn is converted to S-adenosyl methionine (SAM), the global CH3 donor in DNA methylation(Reference Crider, Yang and Berry54). CH3 group deprivation can lead to changes in C1 folate metabolism, metabolites, which can irreversibly perturb DNA methylation and interestingly, induce lesions associated with the pathogenesis of cancer. For instance, Pogribny et al. reported that male F344 rats fed a diet deficient in CH3 groups (low methionine and choline, and folic acid negative) for 9 weeks exhibited a 70 % reduction in SAM when compared with mice on a control diet, while S-adenosyl homocysteine (SAH) was unaffected, thus a significant decrease in the SAM:SAH ratio, an important predictor of methylation capacity, was observed. The CH3-deficient diet also led to a 60 % increase in unmethylated CCGG sites obtained from liver tissue. Interestingly, the reintroduction of a CH3-sufficient diet resulted in normalised DNA methylation in rats who were fed a CH3-deficient diet for 9 weeks. However, in rats fed a CH3-deficient diet for 18, 24 or 36 weeks, the reintroduction of a CH3-sufficient diet could not reverse the hypomethylation induced. Significantly, the appearance of glutathione-S-transferase π, a characteristic of hepatocarcinogenesis was observed despite the reintroduction of the CH3-sufficient diet, even after 9 weeks of exposure to a CH3-deficient diet(Reference Pogribny, Ross and Wise55). Additionally, Jung et al. reported that folic acid for 3 years (800 µg/d) did not influence DNA methylation in moderately hyperhomocysteinaemic Dutch males and females aged 50–70 years(Reference Jung, Smulders and Verhoef56).
In contrast, results from Pufulete et al. suggest that DNA hypomethylation brought about by a low dietary intake of folate could be reversed by folate supplementation. It was reported that a supplement of folic acid for 10 weeks (400 µg/d), in patients with colorectal adenoma, increased serum folate from 7·4 to 13·4 µg/l, and plasma homocysteine decreased 12 %, while DNA methylation increased by 31 and 25 % in leucocytes and colonic mucosa, respectively(Reference Pufulete, Al-Ghnaniem and Khushal57). Interestingly, Park et al. found that folate supplementation produced distinct differences in DNA methylation patterns dependent on body weight. In this study, supplementation of 800 µg/d for 8 weeks in normal weight and obese females aged 18–35 increased serum folate by 86·2 and 109·6 %, respectively. Before supplementation, 10·7 % of CpG sites differed between the different weight categories; this rose to 15·2 % after supplementation. Higher levels of methylation were observed in 52·9 and 55·0 % of obese women before and after treatment, respectively. After treatment, CpG sites were more likely to have reduced levels of methylation; 67·9 and 75·8 % for normal weight and obese females, respectively. Interestingly, while the supplementation-induced methylation changes in genes associated with neural tube closures in women of normal weight, overweight women exhibited changes in methylation in genes associated with folate metabolism, methylation and vitamin B metabolism(Reference Park, Bailey and Shade58).
Conversely, a 3-month 100, 400 and 4000 µg/d supplement of folate resulted in an 11·5, 11·7 and 18·9 % reduction in % 5 CH3-deoxycytidine, respectively, in coagulated blood samples from Chinese women of reproductive age, who showed an average % CH3-deoxycytidine level of 4·42 (sd 0·12) % at enrolment. Interestingly, it was observed that genotype can influence the DNA methylation response to dietary folate. When analysing the effect of a 3-month 4000 µg/d folate supplement in the presence of the methylenetetrahydrofolate reductase 677C→T variant, it was found that there was an 11·6, 18·8 and 19·5 % reduction in % 5 CH3-deoxycytidine for the CC, CT and TT genotypes, respectively, compared with baseline results(Reference Crider, Quinlivan and Berry59).
Caloric restriction
Recent evidence has suggested that caloric restriction (CR), even in the short term, could potentially ameliorate aberrant methylation in disease-associated genes as observed in age-related methylation drift(Reference Kim, Lee and Choi60). Maegawa et al. detailed that CR was able to counteract hypermethylation associated with ageing without producing novel methylation patterns. In both DNA from whole blood of mice and Rhesus monkeys, a significant inverse relationship was observed between CR and the rate of methylation drift. Specifically, there was an average DNA methylation, across twenty-four genes, of 26 (sem 2) and 27 (sem 0·7) % for aged mice and Rhesus monkeys fed ad libitum, compared with 17 (sem 0·7) and 24 (sem 0·9) % in aged mice and Rhesus monkeys, respectively, who underwent CR(Reference Maegawa, Lu and Tahara12). Hahn et al. showed that a 40 % dietary restriction in mice resulted in a reduced number of differentially methylated regions in DNA extracted from the liver. In aged mice fed ad libitum, age differentially methylated 3176 regions, of which 1945 became hypermethylated and 1231 became hypomethylated, whereas aged mice who underwent 40 % dietary restriction exhibited only 2250 differentially methylated regions were identified, of which 1512 became hypermethylated and 738 became hypomethylated(Reference Hahn, Grönke and Stubbs61). To further this, Wang et al. reported that 40 % CR resulted in a predicted 9·4-month reduction in epigenetic age within the livers of 22-month-old mice compared with age-matched controls(Reference Wang, Tsui and Kreisberg62); therefore, it appears that CR may provide a promising treatment strategy for aberrant DNA methylation.
Drug therapy
Due to the association of hypermethylation of promoters and tumour suppressor gene silencing in cancer, the use of drugs that ameliorate this change may provide a successful method for reducing DNA methylation in these regions and enable the re-expression of these genes. Hypomethylating agents 5-azacytidine (azacytidine) or 5-aza-2’-deoxycytidine (decitabine) are two such examples, which were approved for use by the European Medicines Agency in 2008 and 2012, respectively, for patients with myelodysplastic syndromes (MDS).
Following the cellular uptake of decitabine, it is phosphorylated to 5-aza-2’-deoxycytidine-triphosphate, and becomes incorporated into DNA strands in place of cytosines within CpG sites. The substitute nucleotide binds DNMT similarly to cytosine; however, due to the substitution of C5 in the cytosine ring for nitrogen, β-elimination is inhibited and thus covalent bonding is irreversible. This results in enzyme inhibition and eventual degradation of the bound enzyme, and thus a reduction in DNA methylation. Azacytidine acts in a similar way; however, it acts upon RNA. Interestingly, during phosphorylation, approximately 10–20 % is converted to a 5-aza-2′-deoxycytidine-triphosphate precursor and thus acts upon DNA(Reference Stresemann and Lyko63).
While meta-analysis data suggest that both azacitadine and decitabine are superior to best supportive care in patients with MDS(Reference Almasri, Alkhateeb and Damlaj64), there is conflicting evidence on the superiority of these drugs. For instance, Lee et al. conducted a comparative analysis of azacytidine, given for 7 d in a 28 d cycle, and decitabine, given for 5 consecutive days in a 28 d cycle, in patients with MDS, and response rates of 46 and 52 %, a median peak response at 4·2 and 4·0 months, and median survival time of 23·3 and 22·9 months were reported, respectively. While these parameters were not statistically different from one another, it was established that the survival rate was significantly improved in patients >65 taking azacitidine, and patients showed reduced vulnerability to infection, in addition to a lower incidence of grade 3 or 4 of cytopenia(Reference Lee, Kim and Yoon65). Xie et al. conducted a meta-analysis of eleven trials, containing 1392 MDS patients and similarly found that while there was no significant difference between the rate of complete response in patients undertaking decitabine or azacytidine treatment (13 v. 12 %), azacitidine treatment resulted in a significantly higher overall response rate compared with decitabine (73 v. 42 %). Moreover, a statistically significant improvement in overall survival was observed for azacytidine treatment when compared with best supportive care, while no statistical difference was observed for decitabine treatment(Reference Xie, Jiang and Xie66).
In contrast, results from a randomised phase II trial in patients with low–intermediate-risk MDS or chronic myelomonocytic leukaemia indicated an overall response rate of 70 and 49 % for patients who received intravenous decitabine or azacytidine for 3 consecutive days, on a 28 d cycle, respectively. Furthermore, the 1-year event-free survival rate was significantly greater in patients who received decitabine (74 v. 55 %). In addition, haematological improvements were observed in 24 % of patients treated with decitabine compared with 8 % of azacytidine patients, and of the patients who were transfusion dependent at the start of the trial, 32 and 16 % became transfusion independent following decitabine or azacytidine treatment(Reference Jabbour, Short and Montalban-Bravo67).
However, it is important to note the use of such hypomethylating agents should be used with caution due to selectivity concerns. For instance, in one study which used azacytidine to treat the non-invasive breast cancer cell lines MCF-7 and ZR-75-1, the drug lowered DNMT1 and DNMT3b and methylation within the promoters of several pro-metastatic genes, including uPA and MMP2, leading to gene expression. Furthermore, it was shown that the treatment increased the invasiveness of both cell lines(Reference Chik and Szyf68).
Detecting DNA methylation
In recent years, a systems-orientated approach has become common place in bioscience research(Reference Mc Auley and Mooney69–Reference Mc Auley, Guimera and Hodgson73). The essence of this approach is to utilise novel approaches to study molecules, cells or entire organisms. Nutrition research is no different and is beginning to benefit from this new paradigm(Reference Mc Auley, Proctor and Corfe74–Reference Morgan, Mooney and Wilkinson80). It can be argued that electrochemical techniques come under this umbrella of systems techniques. For instance, recently, there has been heightened interest in using electrochemical techniques to detect DNA methylation as it can be a rapid, easy to use and cost-effective solution to many of the challenges posed by more conventional methods and enables the quantitative analysis of complex biochemical systems(Reference Hossain, Mahmudunnabi and Masud81).
There are several techniques that can be employed to analyse DNA methylation, many of which require prior bisulphite conversion, which converts unmethylated cytosines to uracil, while methylated cytosines remain unchanged. These techniques include bisulphite sequencing(Reference Li and Tollefsbol82), methylation-specific PCR(Reference Herman, Graff and Myohanen83), pyrosequencing(Reference Tost and Gut84) and immuno-based recognition(Reference Rauch and Pfeifer85). Methods which do not require prior bisulphite conversion include high-performance liquid chromatography(Reference Armstrong, Bermingham and Bassett86), MS(Reference Lin, Zhang and Liu87), microarray analysis(Reference Schumacher, Kapranov and Kaminsky88), surface plasmon resonance(Reference Sina, Howell and Carrascosa89) and surface-enhanced Raman spectroscopy(Reference Hu and Zhang90). Many of these methods have several drawbacks including the need for expensive laboratory equipment and/or biological molecules coupled with long analysis times and the requirement for highly skilled operators.
An example of an elegant electrochemical DNA-methylation sensor is the eMethylsorb method of Koo et al. The method consists of two steps. First, a gold electrode is exposed to a solution of bisulfite-modified DNA. This exploits the findings of Kimura-Suda et al., who demonstrated that single-stranded homo-oligonucleotides adsorbed onto gold with the following affinity A > C ≥ G > T(Reference Kimura-Suda, Petrovykh and Tarlov91). The DNA adsorption essentially blocks (or passivates) the gold surface, decreasing its reactivity. The lower the methylation level of the original DNA, the higher the number of adenines present in the bisulfite-treated sample. Consequently, unmethylated DNA results in a more passivated and less reactive surface than methylated DNA. In the second step of the eMethylsorb method, the reactivity of the gold electrode surface is measured in an electrochemical reaction. Initially, Koo et al. developed the eMethylsorb method using disposable gold screen-printed electrodes (consisting of a 4 mm diameter gold working electrode, gold counter electrode and silver reference electrode) exposed to the solutions of synthetic oligonucleotides diluted in 5× saline sodium citrate buffer, designed to represent bisulphite-modified and asymmetrically amplified methylated and unmethylated versions of a fifty-three-base section, containing eight CpG sites, of the EN1 gene promoter. After the adsorption step, the reactivity of the modified gold service was measured by performing differential pulse voltammetry in an electrolyte containing 2·5 mm ferrocyanide, 2·5 mm ferricyanide and 100 mm KCl, where the peak current for the reduction of Fe3+ to Fe2+ inversely correlated with the level of DNA adsorption on the gold electrode.
Optimisation of the adsorption step revealed that the greatest current response difference (between methylated and unmethylated samples) was observed when 50 nm of synthetic oligonucleotides were adsorbed for 10 min (in quiescent solution) at pH 7·0. The method was used to successfully detect 10 % methylation in heterogeneous samples of synthetic oligonucleotides. Furthermore, the technique was able to detect 10 % methylation in heterogeneous samples containing various combinations of MCF-7 and whole-genome amplified DNA. Interestingly, the sensitivity of the method was significantly greater for these 140-base DNA samples in comparison to the fifty-three-base synthetic oligonucleotides(Reference Koo, Sina and Carrascosa92).
In a related study, the same research group used a 2 mm gold disk working electrode (Pt counter electrode and Ag/AgCl reference electrode) to detect methylation levels in the same synthetic oligonucleotides (in 5× saline sodium citrate buffer). The electrochemical reactivity of the modified gold surface was measured via differential pulse voltammetry in a solution of 2·5 mm ferrocyanide, 2·5 mm ferricyanide and 10 mm PBS. Using the two-step eMethylsorb procedure, the greatest relative current difference was observed between methylated and unmethylated DNA, when 200 nm DNA was adsorbed for 10 min (in quiescent solution) at pH 7·0. Again a negative linear relationship between % methylation in heterogeneous samples of synthetic methylated and unmethylated oligonucleotides and the relative current response was observed (R 2 0·99 398). Sina et al. also investigated the effect of the number of methylated CpG sites within the fifty-three-base synthetic oligonucleotide (0, 1, 4 and 8). A negative linear relationship was observed between the number of methylated CpG sites and relative differential pulse voltammetry current response (R 2 0·971 411). Finally, it was determined that only 20 µl of secondary PCR product (from real DNA samples) in 200 µl buffer was required to produce a considerable difference in relative current. Once again, the sensitivity of the method greatly improved on moving from synthetic to real DNA samples(Reference Sina, Howell and Carrascosa89).
Our project set out to improve the repeatability and sensitivity of the eMethylsorb method via a new approach to the adsorption step and the electrochemical technique (Fig. 2). The new procedure was optimised using thirty-base synthetic oligonucleotides, containing six CpG sites, designed to represent bisulphite-modified and asymmetrically amplified methylated and unmethylated versions of a region downstream of the transcription site of the EN1 gene promoter(Reference Thompson, Davies and Mc Auley93).
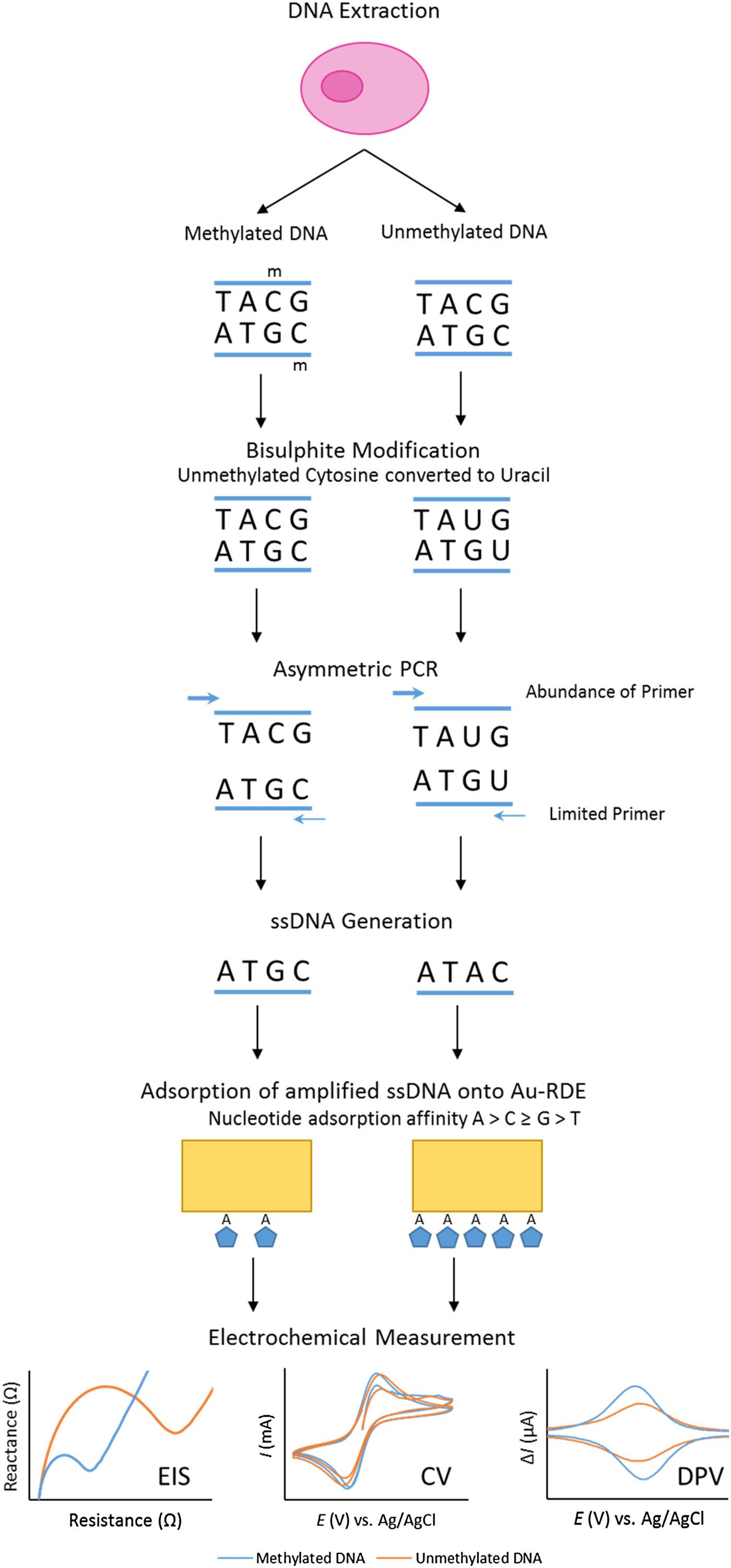
Fig. 2. Overview of bisulphite treatment, asymmetric PCR and electrochemical measurement.
It was also imperative to test if % methylation could be determined using these optimised electrochemical procedures in a heterogeneous sample. This was to reflect biopsy samples gained in a clinical setting, as tumours are often found to contain cells exhibiting diverse phenotypic features; including methylation status. This phenomenon, termed intra-tumour heterogeneity, has been observed in multiple cancers including breast(Reference Moelans, de Groot and Pan94), lung(Reference Quek, Li and Estecio95), endometrial(Reference Varley, Mutch and Edmonston96) and prostate cancer(Reference Litovkin, Van Eynde and Joniau97). To test the applicability of the procedure in detecting methylation in DNA derived from human subjects, the procedure was repeated using bisulphite-modified and asymmetrically amplified 140-base single-stranded DNA from the EN1 region of DNA extracted from the breast cancer cell line MCF-7 (methylated) and whole-genome amplified DNA (unmethylated). Our method successfully detected high methylation levels in the breast cancer cells(Reference Thompson, Davies and Mc Auley93).
Conclusion
This review has highlighted a wide variety of dietary components that can influence DNA methylation status during life. Based on our survey of the literature, it can be argued that many instances of aberrant DNA methylation are the direct result of diet. Nowhere is this more apparent than cancer, because the methylation changes that are a hallmark of many cancers are influenced by dietary factors such as folate consumption, energy intake and polyphenol content. This review has reinforced the idea that identifying DNA methylation changes early could be an effective means of predicting cancer risk. An important take-home message from the present review is that the early detection of cancer could be achieved by monitoring methylation levels within biomarker genes such as the EN1 gene. Finally, this review has revealed that the goal of early cancer detection could be achieved by using novel electrochemical techniques to quantify DNA methylation levels. There is little doubt that techniques such as this will prove to be an invaluable tool in the detection of cancer in the future.
Acknowledgement
A. E. M. was funded by a University of Chester PhD scholarship.
Financial support
This study was partially funded by the Higher Education Funding Council for England Innovation Fund.
Conflicts of interest
None.
Authorship
M. T. M. A. and T. J. D. conceived the project. A. E. M. and M. T. M. A. drafted the manuscript.


