


From an evolutionary standpoint, pain can be considered a necessary evil, providing a potent warning system to protect an individual from present and future harm. However, not all pain is part of this adaptive response, e.g. persistent pain after injury healing (chronic pain) or pain arising from damage to nerve tissue (neuropathic pain). Pain is the most common complaint in those seeking a physician, and a recent study suggests that pain represents the greatest economic burden of any pathological condition in the USA, with an estimated annual cost of $565–635 billion( Reference Gaskin and Richard 1 ). Many chronic pain states are refractory to standard analgesics, and even in those which do respond, pain control can be incomplete or only short-term in nature. Of the imperfect currently available analgesics, the most efficacious are the opioids which exploit an endogenous pain control pathway within the central nervous system. However, opioids have significant issues with tolerance, dependence, respiratory depression and opioid-induced hyperalgesia( Reference Angst and Clark 2 ). Over the past few decades, the existence of a second endogenous anti-nociceptive pathway has been revealed: the endocannabinoid (EC) system.
Cannabinoids and the endocannabinoid system
Extracts from Cannabis sativa have been used as analgesics for centuries, but paucity of knowledge of the molecular pharmacology, combined with moral reservations about the psychotropic effects of this drug and its abuse for recreational purposes, led to its prohibition in the early 20th century. However, the identification of Δ9-tetrahydrocannabinol as the major psychoactive component of cannabis extracts( Reference Mechoulam and Gaoni 3 ), and the subsequent isolation of a cannabinoid receptor highly expressed in nervous tissue( Reference Devane, Dysarz and Johnson 4 – Reference Herkenham, Lynn and Johnson 6 ), led to an explosion of research interest in this area. Since Devane's landmark papers, firstly identifying the cannabinoid receptor( Reference Devane, Dysarz and Johnson 4 ), and subsequently anandamide (AEA) the first endogenous ligand( Reference Devane, Hanus and Breuer 7 ) over 5000 articles featuring EC have been published. The extent of the literature signifies the breadth of pivotal roles this system plays in physiological and pathophysiological functioning, and it is now known that in neurones these are fundamentally predicated on the modulation of neuronal signalling via retrograde inhibition (for reviews, see( Reference Kano, Ohno-Shosaku and Hashimotodani 8 , Reference Katona and Freund 9 )).
The EC system is composed of two G protein-coupled cannabinoid receptors (cannabinoid type 1 receptor (CB1) and cannabinoid type 2 receptor (CB2)), the EC ligands that activate them, and their synthetic and catabolic enzymes. The EC system possesses several unique properties when compared with other neurotransmitter systems, and it is these properties that underlie its role in analgesia: (i) EC act in a retrograde manner at neuronal synapses, being synthesised in the post-synaptic cell and travelling back across the synapse to interact with the receptors on the pre-synaptic cell( Reference Katona, Sperlagh and Sik 10 , Reference Egertova and Elphick 11 ); (ii) EC are not stored in vesicles prior to release, but instead are produced through activity-driven ‘on demand’ synthesis following strong neuronal activation( Reference Ohno-Shosaku, Maejima and Kano 12 , Reference Wilson and Nicoll 13 ); (iii) The EC system involves a multitude of ligands acting at just two major receptors, in contrast to the single ligand/multiple receptor paradigm present in other systems (e.g. glutamate, γ-aminobutyric acid, 5-hydroxytryptamine, etc.)( Reference Di Marzo and De Petrocellis 14 ).
EC are thought to act as a brake on neuronal hyper-activity, being produced in response to high levels of stimulation and feeding back negatively on the circuit through interaction with pre-synaptic cannabinoid receptors. In pain pathways, these actions produce analgesia by inhibiting the transmission of pain signals.
Cannabinoid receptors
The cannabinoid receptors have divergent expression patterns underlying their separate physiological roles. CB1 is predominantly found on nerve cells, while CB2 is mostly expressed on cells of the immune system, with some evidence of limited neuronal expression (see discussion and references in( Reference Rani Sagar, Burston and Woodhams 15 )).
The CB1 receptor is predominantly found pre-synaptically on axon terminals( Reference Katona, Sperlagh and Sik 10 ), and is coupled with adenylate cyclase via Gi/o proteins. Activation leads to a reduction of pre-synaptic neurotransmitter release via inhibition of N- and P/Q-type calcium channels, and activation of potassium channels( Reference Galiegue, Mary and Marchand 16 ). The net result of these actions can be inhibition or excitation of neuronal circuits, depending on whether the pre-synaptic cell secretes excitatory or inhibitory neurotransmitters.
Endocannabinoids
The EC are lipid signalling molecules, produced on-demand by the activity-dependent enzymatic cleavage of membrane phospholipids( Reference Sagar, Gaw and Okine 17 ). The most widely investigated are the arachidonic acid derivatives AEA and 2-arachidonoyl glycerol (2-AG; structures shown in Fig. 1). AEA was the first to be discovered, and is thus the most studied, although it is present at far lower levels in tissue than 2-AG (∼170-fold lower in brain( Reference Stella, Schweitzer and Piomelli 18 )). Both AEA and 2-AG activate cannabinoid receptors; 2-AG is a full agonist at both CB1 and CB2 receptors( Reference Sugiura, Kondo and Kishimoto 19 ), whereas AEA shows slight selectivity for CB1 over CB2. AEA has also been shown to activate the ion channel receptor TRPV1( Reference Zygmunt, Petersson and Andersson 20 ) and as such can be considered an EC/endovanilloid substance (for review, see( Reference Starowicz and Przewlocka 21 )).
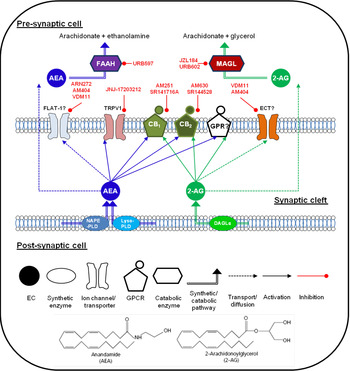
Fig. 1. (Colour online) Endocannabinoid (EC) signalling at a notional neuronal synapse. The major synthetic, signalling and catabolic pathways for anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are shown. Alongside cannabinoid receptors (CB1) and (CB2) the other G protein coupled receptors (GPCR) may be involved in cannabinoid signalling. FLAT-1, a truncated form of fatty acid amide hydrolase (FAAH), and ECT are the putative EC transporters. MAGL, monoacylglycerol lipase; DAGL, diacyglycerol lipase; GPR, specific G protein-coupled receptor such as GPR55. Compounds in red are recognised enzyme inhibitors/receptor antagonists, which can modulate EC signals.
Endocannabinoids and nutrition
The EC are lipid-based, and as such are more susceptible to dietary-induced fluctuations in tissue levels than other transmitter substances( Reference Artmann, Petersen and Hellgren 22 , Reference Watanabe, Doshi and Hamazaki 23 ). Elevations in EC levels in epididymal fat and pancreas have been reported in a mouse model of diet-induced obesity( Reference Matias, Gonthier and Orlando 24 ), and these diet-induced elevations may alter the EC system functionally, since female mice on a high-fat diet show decreased sensitivity to Δ9-tetrahydrocannabinol( Reference Wiley, Jones and Wright 25 ). Recent clinical data suggests dietary modulation of EC also occurs in human subjects( Reference Berge, Piscitelli and Hoem 26 ), with an 84 % decrease in plasma AEA observed following chronic exposure to dietary DHA and EPA. Although the functional relevance of circulating EC levels remains unclear, this robust effect of fatty acid intake suggests that nutritional factors can influence the EC system, and may offer a means to modulate the analgesic potential discussed here.
Endocannabinoid synthesis and degradation
The major EC have distinct synthetic and degradative pathways, with the localisation of the synthetic and degradative machinery for each ligand determining its physiological effects (for review, see( Reference Luchicchi and Pistis 27 )). A schematic diagram of the actions of EC at a generic neuronal synapse can be seen in Fig. 1.
AEA was initially thought to be synthesised by a pathway involving the enzyme N-acyl-phosphatidylethanolamine-hydrolysing phospholipase D( Reference Di Marzo, Fontana and Cadas 28 , Reference Okamoto, Morishita and Tsuboi 29 ). However, generation of an N-acylphosphatidylethanolamine-hydrolysing phospholipase D knock-out mouse revealed normal brain levels of AEA( Reference Leung, Saghatelian and Simon 30 ), and additional synthetic pathways have since been identified( Reference Simon and Cravatt 31 , Reference Simon and Cravatt 32 ). It remains unclear which pathway predominates in the production of AEA in pain pathways. The major catabolic enzyme for AEA is fatty acid amide hydrolase (FAAH), although other catabolic pathways have been identified (for review see( Reference Ueda, Tsuboi and Uyama 33 )).
2-AG is produced from cleavage of membrane phospholipids by phospholipase C-β to produce diacylglycerol species, and subsequent cleavage to form 2-AG by the enzymes diacyglycerol lipase-α and β( Reference Bisogno, Howell and Williams 34 ), and metabolism is primarily via monoacylglycerol lipase (MAGL)( Reference Dinh, Carpenter and Leslie 35 ). Other enzymes participating in 2-AG metabolism have been identified( Reference Blankman, Simon and Cravatt 36 ), although their roles in termination of 2-AG signalling have yet to be fully elucidated.
Pain and nociception
Pain is an integrative experience, involving physiological, emotional and cognitive aspects (for a useful glossary of pain terminology, see( Reference Loeser and Treede 37 )). The subjective experience of pain varies significantly between individuals and cannot be reported by the non-human animals on which the majority of basic pain research is conducted. For the purposes of this review, we differentiate between the subjective experience of pain, and the measurable neuronal events which underlie it, hereafter referred to as nociception.
Nociceptive pathways begin with the transduction of a noxious stimulus, such as mechanical pressure, into action potentials by a specialised class of sensory afferent neurones in the periphery (e.g. mechanoreceptors in the skin). Action potentials travel via the axon of the primary afferent neurone, past the cell body located in a dorsal root ganglion, to a synapse in the superficial dorsal horn of the spinal cord. Following the integration of inputs from multiple cells types within the spinal cord, these action potentials will then pass up one of several ascending pathways to the brainstem, and subsequently to the thalamus, which then relays the signal to higher brain regions involved in the sensory (e.g. the somatosensory cortex) and emotional/affective (e.g. the amygdala and cingulate cortex) aspects of pain. There is significant cross-talk between supra-spinal nociceptive regions, and nociceptive signals can be amplified or dampened by descending modulatory pathways projecting from the brain to the spinal cord (pathways reviewed in( Reference Basbaum, Bautista and Scherrer 38 – Reference Todd 40 )). Fig. 2 displays a schematic of a typical nociceptive pathway.

Fig. 2. (Colour online) Nociceptive pathways. Schematic of nociceptive pathways. Nociceptive stimuli are conducted from the periphery to the dorsal horn of the spinal cord, and then transmitted to the supra-spinal regions via the spinothalamic tract (STT, blue) and spinoparabrachial tract (SPBT, red). The major descending modulatory control pathway (DMCP, purple) is displayed on the right. This pathway crosses the midline at the level of the medulla. Coloured areas indicate the position of synapses in each pathway. The positions of laminae I–VI in the dorsal horn are indicated by dotted lines, while the black region in the brain represents the lateral ventricles. Thal., thalamus; VMH, ventromedial hypothalamus; PbN, parabrachial nucleus; PAG, periaqueductal grey matter; RVM, rostroventral medulla; Pyr., pyramidal tract; DRG, dorsal root ganglion. Adapted from( Reference Hunt and Mantyh 39 ).
Location of the endocannabinoid system in the pain pathway
The components of the EC system are expressed ubiquitously throughout the pain processing pathways, underlining its key modulatory role in nociception. Both ligands and receptors can be detected in the periphery, at the level of the spinal cord, and in nociceptive regions of the brain( Reference Herkenham, Lynn and Johnson 6 , Reference Sagar, Kendall and Chapman 41 – Reference Calignano, La Rana and Giuffrida 71 ). CB1 is predominantly localised in neurones, while CB2 is found in immune cells, although there is some evidence for non-neuronal CB1 in B cells of the immune system( Reference Kaplan 47 – Reference Pacher and Mechoulam 49 ) and astroglial cells of the central nervous system( Reference Salio, Doly and Fischer 59 , Reference Alkaitis, Solorzano and Landry 64 , Reference Stella 68 ). CB2 expression has been reported in microglia of the central nervous system( Reference Racz, Nadal and Alferink 62 – Reference Alkaitis, Solorzano and Landry 64 ), with some evidence suggesting neuronal CB2 expression (see commentary in( Reference Atwood and Mackie 69 )), although this remains controversial.
Peripheral mechanisms
The peripheral compartment of the EC system plays a substantial role in cannabinoid-receptor-mediated anti-nociception, as demonstrated by the greatly reduced efficacy of locally and systemically administered cannabinoids following selective deletion of peripheral CB1 ( Reference Agarwal, Pacher and Tegeder 70 ). Evidence indicates that both neuronal and non-neuronal cannabinoid receptors contribute to the anti-nociceptive effects of peripheral EC, and the contributions of these and the ligands AEA, and 2-AG have been partially revealed via use of specific enzyme inhibitors and receptor antagonists in animal models of pain.
The most studied paradigm is that of inflammatory pain, in which application of an inflammatory substance to the rodent hindpaw elicits an oedemic response and measureable nociceptive behaviour. Peripheral administration of AEA in the formalin model temporarily reduced the nociceptive behaviour in a CB1-sensitive manner( Reference Calignano, La Rana and Giuffrida 71 ). Conversely, blocking CB1 and/or CB2 receptors prior to formalin administration increased nociceptive responses, suggesting an intrinsic role for EC in inflammatory pain. This may be restricted to early inflammatory pain states, since the hindpaw levels of EC are decreased at later time-points (see reviews in( Reference Sagar, Gaw and Okine 17 , Reference Maione, De Petrocellis and de Novellis 72 , Reference Beaulieu, Bisogno and Punwar 73 )). Similarly, intra-plantar administration of 2-AG blocked the second phase of formalin-evoked pain behaviour in rats( Reference Guindon, Desroches and Beaulieu 74 ), via a CB2-mediated mechanism.
Exogenously administered AEA and 2-AG are rapidly metabolised by FAAH and MAGL, respectively. Further studies have therefore focused on the use of FAAH and MAGL inhibitors to prolong the effects of endogenous EC actions. Systemic inactivation of FAAH via compounds such as URB597, OL135 and PF-3845 has been shown to be anti-nociceptive in models of acute and inflammatory pain( Reference Jayamanne, Greenwood and Mitchell 75 – Reference Russo, Loverme and La Rana 80 ). Elevations in both AEA and 2-AG have been shown, as well as reduced carrageenan-induced hyperalgesia( Reference Jhaveri, Richardson and Robinson 81 ) and expansion of peripheral receptive field size of wide dynamic range neurons (a marker of central sensitisation) following intra-plantar URB597( Reference Sagar, Kendall and Chapman 82 ). Similarly, capsaicin-induced pain behaviour and thermal hypersensitivity were attenuated following blockade of peripheral MAGL via JZL184( Reference Spradley, Guindon and Hohmann 83 ), and peripheral administration of JZL184 produces local inhibition of MAGL activity, increased levels of 2-AG and anti-nociceptive effects in both phases of the formalin model through mechanisms involving both CB1 and CB2 ( Reference Guindon, Guijarro and Piomelli 84 ). A peripherally restricted FAAH inhibitor URB937( Reference Clapper, Moreno-Sanz and Russo 85 ), which cannot cross the blood–brain barrier, blocked the hypersensitivity induced by both inflammation and peripheral nerve injury via a CB1-mediated mechanism, without altering thermal nociceptive thresholds. These data reinforce the notion that peripheral EC tone is increased in pain states, and that actions of EC at peripheral CB1 receptors can reduce the transmission of nociceptive information. Further enhancing EC signalling may therefore prove an effective analgesic strategy, although it should be noted that FAAH and MAGL are not the only enzymes that can degrade EC( Reference Ueda, Tsuboi and Uyama 33 , Reference Blankman, Simon and Cravatt 36 ), and thus, the effects of specific enzyme inhibitors may not present the entire story.
Alongside direct anti-nociceptive effects at CB1 receptors on afferent neurones, EC can also act at peripheral CB2 on immune cells such as macrophages( Reference Han, Lim and Ryu 86 ), lymphocytes( Reference Cencioni, Chiurchiăą and Catanzaro 87 ) and mast cells( Reference Samson, Small-Howard and Shimoda 88 ). CB2 receptor activation inhibits the production and release of pro-inflammatory and pro-nociceptive mediators, such as reactive oxygen species( Reference Hao, Jiang and Fang 89 ) and cytokines( Reference Cencioni, Chiurchiăą and Catanzaro 87 ). In addition, metabolism of 2-AG produces arachidonic acid, a key precursor of pro-inflammatory PG, and disruption of 2-AG hydrolysis reduces the available pool of arachidonic acid and thus reduces inflammation( Reference Nomura, Morrison and Blankman 90 ). In summary, elevating levels of peripheral AEA produces anti-nociceptive effects in models of inflammatory and neuropathic pain, largely via the inhibitory actions of CB1 receptors on the primary neurones which transmit nociceptive signals. Similarly, increased 2-AG signalling at peripheral CB1 receptors is also anti-nociceptive, but there also appears to be a prominent CB2-mediated component, probably involving the inhibition of pro-nociceptive actions of immune cells.
Spinal mechanisms
Exogenous application of EC is anti-nociceptive at the level of the spinal cord( Reference Starowicz, Makuch and Osikowicz 91 , Reference Welch, Huffman and Lowe 92 ), while intra-thecal administration of a CB1 receptor antagonist produces hyperalgesia in mice( Reference Richardson, Aanonsen and Hargreaves 93 ), enhancing nociception-evoked firing of wide dynamic range neurones in the dorsal horn of the spinal cord( Reference Chapman 94 ). In addition, spinal EC are elevated in animal models of acute and chronic pain( Reference Sagar, Jhaveri and Richardson 95 – Reference Okine, Norris and Woodhams 97 ). These data indicate a role for the spinal EC system in nociceptive transmission. In the surgical incision model of acute resolving pain in rats, spinal levels of AEA are reduced at early time-points coinciding with maximal mechanical hypersensitivity, returning to baseline as nociceptive behaviour subsides( Reference Alkaitis, Solorzano and Landry 64 ). In comparison, 2-AG levels were elevated at time-points coinciding with the appearance of glial cell activation and up-regulation of CB2 receptors, suggesting a temporal segregation of AEA and 2-AG signalling. In agreement with these data, spinal levels of AEA are significantly elevated at early time-points in the chronic constriction injury (neuropathic pain model) model of neuropathic pain in mice( Reference Petrosino, Palazzo and de Novellis 98 ). Spinal administration of URB597 reduced mechanically evoked responses of wide dynamic range neurones in rats that underwent spinal nerve ligation and this effect was blocked by pre-administration of a CB1 selective receptor inverse agonist/antagonist( Reference Jhaveri, Richardson and Kendall 99 ). Similarly, we have shown that spinal application of the MAGL selective inhibitor JZL184 produced a dose-related reduction in mechanically-evoked nociceptive neurotransmission in the spinal cord of naive rats, which was reversed in the presence of the CB1 selective antagonist, a CB1 selective receptor inverse agonist/antagonist( Reference Woodhams, Wong and Barrett 100 ). Spinally administered JZL184 was also able to prevent intra-plantar carrageenan-induced receptive field expansion of dorsal horn wide dynamic ranges, indicating that the inhibition of MAGL can block mechanisms underlying the development of central sensitisation following peripheral inflammation.
Further evidence of the involvement of the spinal EC system is provided by alterations seen in receptor expression in established pain states. CB1 expression is elevated in the spinal cord of neuropathic rats from 4 d post-injury, with levels continuing to rise until day 14( Reference Lim, Sung and Ji 101 ) while CB2 receptor up-regulation also occurs by day 4( Reference Zhang, Hoffert and Vu 102 ). Genetic deletion of CB2 receptors results in enhanced microglia and astrocytic expression in the contralateral spinal horn following nerve injury, accompanied by profound contralateral mechanical and thermal allodynia( Reference Racz, Nadal and Alferink 62 ). Conversely, overexpression of CB2 receptors protected against nerve injury-induced thermal and mechanical allodynia and prevented glial activation in the spinal cord. Numerous other studies have also implicated EC signalling in glial cell activation( Reference Alkaitis, Solorzano and Landry 64 , Reference Correa, Docagne and Mestre 103 , Reference Stella 104 ).
Exogenous 2-AG has been shown to stimulate microglial migration, whereas the CB2 receptor antagonist a CB2 selective receptor antagonist inhibits basal microglial migration( Reference Walter, Franklin and Witting 105 ). However, caution should be taken when interpreting these results as cell culture models might not reflect the in vivo situation. Nevertheless, these converging lines of research strongly suggest that the EC system, especially at the level of the spinal cord, is intimately involved in glial cell signalling. Further information regarding the links between the EC system and neuro-glial interactions can be found in the following review( Reference Bilkei-Gorzo 106 ). The analgesic actions of EC and cannabinoid receptor activation in the spinal cord suggest that targeting the EC system at this level could inhibit both neuronal hyper-excitability and glial cell activation. Thus, enhancing this endogenous pathway could prove to have a wide range of therapeutic applications in the treatment of multiple pain sates, including the underlying central sensitisation.
Supra-spinal mechanisms
The supra-spinal sites of cannabinoid anti-nociceptive action were first identified in rodents via microinjection of CB1 ligands into pain-associated regions including the rostroventral medulla, the dorsal raphe nucleus, the periaqueductal grey matter and the amygdala, prior to tests of acute nociception( Reference Martin, Patrick and Coffin 107 ). Later work revealed the role of endogenous ligands by demonstrating mobilisation of AEA( Reference Walker, Huang and Strangman 108 ), and CB1-mediated anti-nociception, following either electrical stimulation of these regions or a peripheral administration of formalin( Reference Rea, Roche and Finn 109 ). Enhancing AEA signalling in these areas via inhibition of FAAH activity is anti-nociceptive in acute pain( Reference Maione, Bisogno and de Novellis 110 ), probably via disinhibition of descending inhibitory inputs from the brainstem to the spinal cord, inhibiting spinal nociceptive signalling (for a review of the pathways involved, see( Reference Basbaum, Bautista and Scherrer 38 – Reference Todd 40 )).
Supra-spinal EC are also responsible for a phenomenon known as stress-induced analgesia, in which brief exposure to environmental stress (e.g. immersion in cold water, or an electric shock to the paw) produces reduced nociceptive responses in a subsequent pain test. Detailed study of this effect revealed the mobilisation of both AEA and 2-AG in the periaqueductal grey matter( Reference Hohmann, Suplita and Bolton 111 ), and suggested that 2-AG acting at CB1 receptors was the predominant mechanism. Additional work indicated that further enhancing stress-induced EC signalling via FAAH or MAGL inhibition produces still greater anti-nociceptive effects( Reference Suplita, Farthing and Gutierrez 112 , Reference Suplita, Gutierrez and Fegley 113 ), and confirmed the pivotal role of 2-AG signalling in the periaqueductal grey matter( Reference Gregg, Jung and Spradley 114 ). The involvement of stress in human pain responses and the presence of an EC-mediated mechanism are now being studied clinically( Reference Vachon-Presseau, Martel and Roy 115 ). These data indicate a physiological role for supra-spinal EC in acute nociception, although agonism of supra-spinal CB1 receptors is an unappealing prospect due to the psychotropic side-effects. Instead, research has focused on modulating the altered EC signalling seen in pain states.
The EC system is plastic, with changes in levels of receptor expression, ligand concentrations and synthetic and catabolic enzymatic activity occurring in pain states (reviewed in( Reference Rani Sagar, Burston and Woodhams 116 )). Elevated levels of EC in the periaqueductal grey matter and dorsal raphe nucleus have been observed at 3 and 7 d in the chronic constriction injury model of neuropathic pain( Reference Petrosino, Palazzo and de Novellis 98 ). Interestingly, desensitisation of CB1 receptors in a pain-related cortical brain region has been described in this model at 10 d, when nociceptive behaviour is maximal( Reference Hoot, Sim-Selley and Poklis 117 ). CB1 receptor desensitisation is known to occur following chronic exposure to ligands( Reference Sim-Selley 118 ), and thus may reflect the result of chronically elevated EC levels in the brain. It remains to be seen whether pharmacologically enhancing EC signalling in pain-related brain regions is advantageous or detrimental under these conditions.
Supra-spinal EC can also modulate the affective (or emotional) aspects of pain, which are mediated by frontal and limbic brain regions, and can be dissected from the somatosensory aspects. A recent neuroimaging study of the analgesic effects of Δ9-tetrahydrocannabinol on capsaicin-evoked cutaneous pain in human subjects revealed no change in the intensity of pain sensation( Reference Lee, Ploner and Wiech 119 ). Instead, test subjects reported that Δ9-tetrahydrocannabinol reduced the unpleasantness of capsaicin-evoked cutaneous ongoing pain, rather than reduced sensation, concurrent with reduced activity in the anterior cingulate cortex and enhanced activity in the right amygdala compared with the control subjects. Whether modulation of the EC system can mimic these effects is as yet unknown. Ablation of the anterior cingulate cortex has been utilised as an effective last-resort treatment for intractable pain( Reference Foltz and White 120 ), and thus unlocking the potential of the EC system in this region could address the great unmet clinical need in chronic pain states.
Novel metabolic pathways
Modulating EC levels by FAAH and MAGL inhibition, as described earlier, can produce anti-nociceptive effects. However, bioactive lipids such as AEA and 2-AG are promiscuous, and can be metabolised by multiple enzymes( Reference Alexander and Kendall 121 ). Artificially elevating EC levels can unmask alternative metabolic routes, producing additional bioactive products. Interestingly, pathological conditions such as chronic pain states are associated with changes in levels of enzymes, such as cyclooxygenase (COX), lipoxygenase, αβ hydrolase and members of the cytochrome P450 family( Reference Fowler 122 – Reference Snider, Nast and Tesmer 125 ), which can metabolise the EC to novel lipid signalling molecules. The physiological actions of these metabolic products are as yet unknown, but some preliminary investigations have been performed. The cytochrome P450 metabolite of AEA, 5,6-epoxyeicosatrienoic acid ethanolamide, has been shown to be a potent CB2 receptor ligand( Reference Snider, Nast and Tesmer 125 ), whereas the 15-lipoxygenase metabolite of 2-AG, 15-hydroxyeicosatetraenoic acid glyceryl ester, acts as a PPARα agonist( Reference Kozak, Crews and Morrow 123 ). COX-2 metabolites of AEA and 2-AG have been shown to have pro-nociceptive actions in the spinal cord. COX-2 metabolises AEA to prostamide F2α, whose spinal application increases the firing of nociceptive neurons and reduces paw withdrawal latencies, and levels of prostamide F2α are elevated in spinal cord tissue in mice with knee inflammation( Reference Gatta, Piscitelli and Giordano 124 ). Similarly, the COX-2 metabolite of 2-AG, PGE2 glycerol ester, is endogenously generated in rat tissue, and induces mechanical allodynia and thermal hyperalgesia following intraplantar administration( Reference Hu, Bradshaw and Chen 126 ). Based on these reports, it is clear that determining the levels of these potential ligands in pain states is of great interest, as many of these metabolites may have effects on pain processing. These findings may also limit the utility of FAAH and MAGL inhibitors as therapeutics in chronic pain states, as they may increase substrate levels for generation of alterative pro-nociceptive EC metabolites, and thus counteract the anti-nociceptive effects of AEA and 2-AG.
Cannabinoids and endocannabinoids in clinical trials
Despite the growing use of medicinal marijuana, and the development of licensed cannabinoid drugs such as Sativex for multiple sclerosis( Reference Garcia-Merino 127 ), concerns remain overdependence, tolerance and the cognitive side-effects produced by these medications. Despite the wealth of pre-clinical data on alternative EC-mediated compounds, the only major clinical trial conducted utilising an EC-directed compound looked at the ability of the selective FAAH inhibitor PF-04457845 to produce analgesia in an osteoarthritic patient population( Reference Huggins, Smart and Langman 128 ). Despite significant elevations in plasma AEA, no analgesic effect was observed. Although disappointing, perhaps, the negative outcome of this trial may indicate a limitation of elevating AEA to induce analgesia in pain sates. Previous work has shown that, in addition to being a CB1 receptor agonist, at higher concentrations AEA binds to and activates the pro-nociceptive TRPV1 channel( Reference Ross 129 ). Since AEA can be also converted into pro-nociceptive signalling molecules in the presence of COX-2 activity, then it is feasible that under pathological pain states where COX-2 activity is up-regulated (e.g. osteoarthritis), pro-nociceptive effects of AEA at TRPV1 may outweigh the anti-nociceptive actions. It has previously been suggested that the development of dual FAAH and COX-2 inhibitors( Reference Fowler, Bjorklund and Lichtman 130 ) or substrate-selective inhibitors of COX-2( Reference Windsor, Hermanson and Kingsley 131 ) would be advantageous in terms of uncoupling the pro- and anti-nociceptive actions of AEA, and producing compounds with superior analgesic profiles. An alternative approach that has been explored pre-clinically utilises N-arachidonoyl-serotonin, a dual FAAH inhibitor/TRPV1 antagonist. This compound has enhanced anti-nociceptive effects v. a FAAH inhibitor alone, in several models of pain( Reference Maione, De Petrocellis and de Novellis 72 , Reference Costa, Bettoni and Petrosino 132 , Reference de Novellis, Palazzo and Rossi 133 ). In addition, changes in AEA biotransformation in the aged patient population may contribute to the lack of analgesia following FAAH inhibition. A recent report describes greater susceptibility to chronic pain and decreased AEA-mediated anti-nociceptive effects in aged animals( Reference Bishay, Haussler and Lim 134 ).
An approach with potentially greater therapeutic appeal than FAAH inhibition involves the targeting of 2-AG signalling. The recent development of MAGL inhibitors such as JZL184 and KML 29( Reference Long, Li and Booker 135 , Reference Chang Jae, Niphakis Micah and Lum Kenneth 136 ), and subsequent preclinical studies suggest that low doses of MAGL inhibitors are devoid of analgesic tolerance( Reference Kinsey, Wise and Ramesh 137 ), and also decrease arachidonic acid pools that are required for the generation of pro-nociceptive molecules such as PGE2( Reference Nomura, Morrison and Blankman 90 ). This approach thus delivers a dual analgesic mechanism, elevating 2-AG and reducing pro-inflammatory PG levels. This may have particular utility in the treatment of inflammatory bowel diseases and the associated pain, especially as MAGL inhibitors have already been shown to attenuate non-steroidal anti-inflammatory drug-induced gastric haemorrhages in mice( Reference Kinsey, Nomura and O'Neal 138 ). Inflammatory bowel diseases, such as irritable bowel syndrome, result in significant pain and distress. Recently, several converging lines of evidence suggest that targeting the EC system may provide much sought after disease modifying therapeutics for these conditions( Reference Alhouayek, Lambert and Delzenne 139 ). At the present stage, little clinical work has been conducted to evaluate the efficacy of FAAH and/or MAGL inhibition in treating inflammatory bowel diseases and associated pain. Given the large unmet clinical need in this area, the disease-modifying potential of an EC therapy with a dual mechanism involving both CB1- and CB2-mediated analgesia, as well as a reduction of pathologically elevated levels of pro-inflammatory mediators, is very appealing.
Concluding remarks
Owing to the breadth and depth of the literature cited here, we have only presented a fraction of the excellent studies conducted in this area, though we hope this information is sufficient to demonstrate the significant potential of targeting the EC system to analgesic effect. It is our belief that such an approach can produce novel efficacious analgesic agents required to help fill the unmet clinical need in chronic pain states. However, to reach this goal, the current gap between the wealth of pre-clinical data and the paucity of clinical trials must be bridged. Translation from the laboratory to the clinic is fraught with difficulties, as evidenced by the failure of the FAAH inhibitor PF-04457845 in osteoarthritic patients, despite a seemingly well-designed study. Future attempts in this area should perhaps utilise patient stratification based on aspects of disease aetiology or stratification based on neuroimaging of the pain matrix in human subjects. Clinical work is also clearly needed that focuses on whether targeting the EC system in highly inflammatory conditions such as irritable bowel syndrome, may offer new analgesic treatments.
It remains to be seen whether medications producing chronic elevations of EC will suffer from similar side-effects to those seen with phytocannabinoids, or the issues of tolerance highlighted in recent animal studies. However, the recent success of Sativex clearly highlights the potential of this area of research.
Acknowledgements
We would like to thank Dr Devi Sagar, Professor Dave Kendall and Professor Victoria Chapman for helpful discussions and advice during the preparation of this work.
Financial support
J. J. B. is funded by Arthritis Research UK Pain Centre funding (grant no. 18769).
Conflicts of interest
None.
Authorship
Both the authors contributed equally to the work.


