


- ARC
arcuate nucleus
- GIP
gastric inhibitory polypeptide
- GLP-1
glucagon-like peptide-1
- PYY
peptide YY
Obesity is a major health problem that is associated with increased morbidity and mortality(Reference Visscher and Seidell1). Its personal, social and economic consequences can be devastating(Reference Visscher and Seidell1–Reference Hedley, Ogden and Johnson3). Substantial research efforts are being directed towards the development of successful weight-loss therapies. Consequently, the understanding of neuroendocrine regulation of food intake and weight gain, especially in relation to the role of gut hormones, has substantially increased over recent years, but new therapies are still awaited(Reference Kaplan4–Reference Yanovski and Yanovski6). Current anti-obesity drugs are moderately effective at achieving weight loss, but considerable adverse effects can occur. Presently, the only effective treatment with a proven mortality benefit is bariatric surgery(Reference Adams, Gress and Smith7, Reference Sjostrom, Narbro and Sjostrom8). The mechanisms underlying the effectiveness of these surgical techniques are not completely understood but alterations in circulating gut hormone levels have been shown to be an important factor(Reference Borg, le Roux and Ghatei9–Reference Naslund, Gryback and Hellstrom11). The gut–brain axis refers in part to gut hormones communicating information from the gastrointestinal tract to the appetite centres within the central nervous system. Changes in these hormones following bariatric surgery may partly explain the mechanism by which surgery reduces appetite and sustains weight loss.
Bariatric surgery
Bariatric surgery, also known as weight-loss surgery, refers to the various surgical procedures performed to treat obesity by modification of the gastrointestinal tract in order to reduce nutrient intake and/or absorption. Procedures for surgical removal of body fat such as liposuction or abdominoplasty are not considered bariatric surgical procedures. Patients who have a BMI ≥35 kg/m2 with an obesity-related comorbidity or patients with a BMI ≥40 kg/m2 who have instituted an adequate exercise and diet programme (with or without adjunctive drug therapy) that has failed meet the National Institute of Clinical Excellence criteria for bariatric surgery(12). Surgical procedures can be grouped in two main categories: restrictive procedures, e.g. gastric banding (Fig. 1); bypass procedures, e.g. Roux-en-Y gastric bypass (Fig. 2). Restrictive surgery works by reducing the volume of the stomach and physically preventing excessive consumption of food(Reference Dixon, Dixon and O'Brien13). However, the most common form of bariatric surgery worldwide is Roux-en-Y gastric bypass surgery(Reference Buchwald, Husemann and Leutenegger14, Reference Buchwald15). Here, a small stomach pouch is created with a stapler device and connected to the distal small intestine. The upper part of the small intestine is then re-attached in a ‘Y’-shaped configuration (Fig. 2). In general, the bypass procedures lead to more weight loss than the restrictive procedures(Reference Sjostrom, Narbro and Sjostrom8). Typically, gastric banding results in a weight loss of approximately 20%, whilst the Roux-en-Y gastric bypass results in approximately 30% weight loss(Reference Maggard, Shugarman and Suttorp16). Weight loss after bypass-type procedures has been shown to be a result of energy intake rather than malabsorption(Reference Pilkington, Gazet and Ang17). Several recent studies have reported a dramatic improvement in obesity-related comorbidities and a decrease in mortality after bariatric surgery(Reference Sjostrom, Narbro and Sjostrom8, Reference Adams, Gress and Smith18, Reference Sjostrom, Lindroos and Peltonen19). Adverse effects after gastric bypass include dumping syndrome in about 20% of patients, leaks at the surgical anastomosis (12%), incisional hernia (7%), infections (6%), deep-vein thrombosis (1–3%)(Reference Ojo, Asiyanbola and Valin20), pulmonary embolism (2%)(Reference Westling, Bergqvist and Bostrom21) and pneumonia (4%)(Reference Encinosa, Bernard and Chen22). To reduce the incidence of complications, patients should be cared for in high-volume centres with clinicians experienced in bariatric surgery(Reference Flum, Salem and Elrod23).
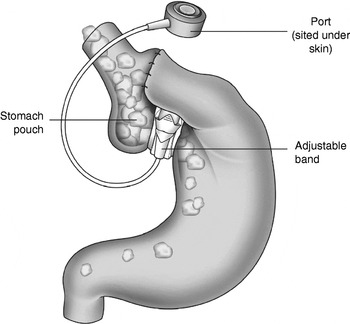
Fig. 1. Gastric banding, a restrictive procedure performed to treat obesity.

Fig. 2. Roux-en-Y gastric bypass, a bypass procedure performed to treat obesity.
Appetite regulation via the gut–brain axis
The hypothalamus contains part of the central melanocortin system and plays a critical role in the regulation of food intake. It has a number of nuclei, including the arcuate nucleus (ARC), paraventricular nucleus, ventromedial nucleus and the dorsomedial nucleus, all of which are interconnected by circuits that regulate energy homeostasis(Reference Wren, Seal and Cohen24). The ARC receives and acts on circulating appetite signals including the modulated release of several key amino acid neurotransmitters(Reference Cone, Cowley and Butler25, Reference Schwartz, Woods and Porte26). The neurons in the medial ARC co-express neuropeptide Y and agouti-related peptide, which stimulate food intake and weight gain by increasing appetite(Reference Schwartz, Woods and Porte26). By contrast, the neurons in the lateral ARC co-express pro-opiomelanocortin (also known as corticotrophin–lipotropin) and cocaine-and-amphetamine-regulated transcript, which both promote weight loss by decreasing appetite(Reference Cone, Cowley and Butler25). Both the ARC and the brainstem are ideally positioned to interact with circulating humoral factors and to receive signals from the periphery(Reference Schwartz, Woods and Porte26). Thus, gut hormones may act directly in the brain after being released into the circulation and entering through the circumventricular organs. Neuropeptide Y can suppress appetite and is a selective ligand for the Y4 receptor subtype, which is expressed at the area postrema and the other appetite-regulating areas of the melanocortin pathway(Reference Balasubramaniam, Mullins and Lin27, Reference Larsen and Kristensen28). The balance between the activities of neuropeptide Y–pro-opiomelanocortin neuronal circuits is critical for the maintenance of body weight(Reference Cone, Cowley and Butler25, Reference Schwartz, Woods and Porte26, Reference Flier29). After food is ingested sensory input to the central nervous system is forwarded by vagal and somatosensory afferent fibres in the gastrointestinal tract that all end in the nucleus tractus solitarius within the brainstem. Reciprocal pathways between the hypothalamus and brainstem pass on information about energy stores and recent food intake, influencing the perception of satiety(Reference Schwartz, Woods and Porte26). These brain centres can respond independently to peripheral signals when communication with higher brain centres is surgically interrupted(Reference Grill and Smith30). Peripheral feedback to the hypothalamus is complex. Many circulating signals, including gut hormones, can have direct access to the ARC(Reference Flier29). These neuronal interactions through central melanocortin pathways therefore reveal the critical role this system has in the regulation of hunger, satiety and energy expenditure(Reference Ellacott, Halatchev and Cone31). However, the homeostatic melanocortin system may protect against weight loss more robustly than it does against weight gain(Reference Schwartz, Woods and Seeley32). In case of changes in body adiposity, the brain triggers physiological mechanisms that resist weight change through compensatory changes in appetite and metabolic rate(Reference Leibel, Rosenbaum and Hirsch33, Reference Brady, Smith and Gold34).
Gut hormones
Ghrelin
Ghrelin is a twenty-eight-amino acid gut peptide derived predominantly from the stomach and pituitary gland(Reference Murakami, Hayashida and Kuroiwa35). So far, it is the only gut hormone with an orexigenic action. It acts via the growth hormone secretagogue receptor to increase food intake in rodents(Reference Wren, Small and Abbott36) and also stimulate food intake in human subjects(Reference Wren, Seal and Cohen24). Clinical studies have thus concentrated on its use as an orexigenic agent in conditions characterized by anorexia and cachexia(Reference Nagaya, Moriya and Yasumura37–Reference Neary, Small and Wren39). Circulating ghrelin levels peak in the fasting state and fall after a meal(Reference Cummings, Purnell and Frayo40). Energy intake seems to be the primary regulator of plasma ghrelin levels(Reference le Roux, Neary and Halsey41). Ghrelin stimulates appetite and food intake also in obese individuals(Reference Druce, Wren and Park42). Ghrelin levels are lower in weight-stable obese individuals and rise after diet-induced weight loss(Reference Cummings, Weigle and Frayo43). The postprandial decrease in plasma ghrelin is absent or attenuated in the obese, which suggests that ghrelin might be involved in the pathophysiology of obesity(Reference English, Ghatei and Malik44, Reference le Roux, Patterson and Vincent45).
Glucagon-like peptide-1
Glucagon-like peptide-1 (GLP-1) is a neuropeptide hormone produced by post-translational processing of the preproglucagon gene in the central nervous system and the gastrointestinal tract(Reference Holst46). Preproglucagon is secreted in the gastrointestinal tract by the endocrine L-cells that also secrete peptide YY (PYY)(Reference Holst46). The GLP-1 receptor belongs to the G-protein-coupled receptors(Reference Larsen, Tang-Christensen and Jessop47). These receptors have been identified in neurons of the nucleus tractus solitarius, extending to regions of the hypothalamus that are important for the regulation of food intake(Reference Crawley and Beinfeld48). Peripheral as well as central GLP-1 administration activates neurons in the ARC, the hypothalamic paraventricular nucleus, the nucleus tractus solitarius and the area postrema, inducing increased satiety and decreased hunger(Reference Larsen, Tang-Christensen and Jessop47, Reference Abbott, Monteiro and Small49). Usually, GLP-1 is released after energy intake, but differences have been observed between normal-weight and obese individuals(Reference Feinle, Chapman and Wishart50–Reference Verdich, Toubro and Buemann52). GLP-1 is a potent incretin. It also suppresses gastric acid secretion and delays gastric emptying(Reference Edwards, Todd and Mahmoudi53, Reference Kreymann, Williams and Ghatei54). These effects can be resolved by vagotomy, indicating an important role of the vagus nerve in mediating the anorectic effects of GLP-1(Reference Abbott, Monteiro and Small49). Peripheral GLP-1 infusions have been found to cause a dose-dependent reduction in food intake, while administration of exenatide (an agonist of the GLP-1 receptor) markedly reduces food intake(Reference Edwards, Stanley and Davis55, Reference Gutzwiller, Goke and Drewe56). Central actions of GLP-1 might also lead to increased energy expenditure by raising body temperature(Reference O'Shea, Gunn and Chen57, Reference Turton, O'Shea and Gunn58). GLP-1 has been shown to promote lipolysis(Reference Ruiz-Grande, Alarcon and Merida59, Reference Villanueva-Penacarrillo, Marquez and Gonzalez60), although some studies have suggested a role in lipogenesis(Reference Villanueva-Penacarrillo, Marquez and Gonzalez60). Glycaemic control in patients with type 2 diabetes mellitus improves after 3 weeks of treatment with subcutaneous GLP-1(Reference Todd, Edwards and Ghatei61), while the agonist exenatide improves HbA1c in the long term(Reference Guerci and Martin62). Furthermore, GLP-1 has been shown to up regulate the expression of pancreatic β-cell genes, promoting β-cell proliferation and inhibiting apoptosis(Reference Soltani, Kumar and Glinka63). Exenatide enhances insulin secretion and suppresses glucagon release(Reference DeFronzo, Ratner and Han64). In phase III clinical trials exenatide has been found to reduce body weight by 3–4 kg, although not all patients respond equally(Reference DeFronzo, Ratner and Han64, Reference Kendall, Riddle and Rosenstock65). Exenatide is not currently approved as an obesity treatment but has been approved for the treatment of type 2 diabetes mellitus. However, nausea is a common adverse effect of this treatment and this effect may relate to reduced gastric emptying or direct effects of the central nervous system(Reference Kendall, Riddle and Rosenstock65).
Peptide YY
As a thirty-six-amino acid peptide PYY is a member of the pancreatic polypeptide family(Reference Tatemoto and Mutt66). It is found throughout the human small intestine, with highest levels in the colon and rectum(Reference Adrian, Ferri and Bacarese-Hamilton67). PYY is released after a meal from the endocrine L-cells of the gastrointestinal tract, where it is co-stored with GLP-1(Reference Adrian, Ferri and Bacarese-Hamilton67, Reference Ali-Rachedi, Varndell and Adrian68). PYY is secreted in proportion to the amount of energy ingested and is independent of gastric distension(Reference Adrian, Ferri and Bacarese-Hamilton67). PYY inhibits gastric, pancreatic and intestinal secretion as well as gastrointestinal motility(Reference Adrian, Savage and Sagor69, Reference Allen, Fitzpatrick and Yeats70). The major form of circulating PYY is the N-terminally truncated PYY3–36, which has high affinity for the Y2 receptor and a lesser affinity for Y1 and Y5 receptors(Reference le Roux, Batterham and Aylwin71). Although initially controversial, peripheral administration of PYY3–36 at physiological doses has now been accepted to reduce food intake in rodents, primates and human subjects in the short term(Reference Chelikani, Haver and Reidelberger72–Reference Tschop, Castaneda and Joost75). PYY-knock-out mice are characterized by dysregulation of energy homeostasis(Reference Boey, Lin and Karl76). PYY3–36 activates anorectic pro-opiomelanocortin-expressing neurons in the ARC and direct intra-ARC administration of PYY3–36 reduces food intake in rats(Reference Batterham, Cowley and Small77). Furthermore, it inhibits neuropeptide Y neurons, which might also contribute to its anorectic effects(Reference Acuna-Goycolea and van den Pol78). These effects of PYY3–36 can be blocked by the administration of a specific Y2 antagonist. In addition, PYY3–36 does not reduce appetite in Y2-knock-out mice(Reference Batterham, Cowley and Small77, Reference Abbott, Small and Kennedy79). Similar to GLP-1, ablation of the vagus–brainstem–hypothalamus pathway leads to a moderation of the anorectic effects, indicating a role of the vagus nerve in the neuronal messaging of PYY(Reference Abbott, Monteiro and Small49). Obese individuals are sensitive to the effects of PYY, as peripheral PYY administration in the obese reduces food intake to the same extent as in normal-weight individuals(Reference Batterham, Cohen and Ellis80), but circulating postprandial PYY levels are lower in the obese(Reference Batterham, Cohen and Ellis80). Exogenous administration of PYY3–36 has attracted considerable interest as a possible therapeutic strategy(Reference White, Dhillo and Liu81). Long-term augmentation of dietary protein induces an increase in plasma PYY levels in mice, leading to less food intake and reduced adiposity(Reference Batterham, Heffron and Kapoor82). PYY3–36 administration in human subjects to levels within the physiological range reduces food intake without causing nausea(Reference Batterham, Cowley and Small77, Reference Batterham, Cohen and Ellis80), whereas higher pharmacological doses can result in nausea(Reference Degen, Oesch and Casanova73). Sensations of hunger, satiety and nausea might all be points along the same physiological spectrum(Reference Greenough, Cole and Lewis83), and nausea is associated with all high-dose satiety-inducing gastrointestinal hormones, including cholecystokinin(Reference Greenough, Cole and Lewis83), oxyntomodulin(Reference Soltani, Kumar and Glinka63) and GLP-1(Reference Naslund, King and Mansten84). Elevated fasting levels of PYY have also been observed in several gastrointestinal diseases associated with appetite loss, including inflammatory bowel disease, steatorrhoea as a result of small intestinal mucosal atrophy and chronic destructive pancreatitis(Reference Adrian, Savage and Bacarese-Hamilton85). Furthermore, in healthy elderly individuals high cholecystokinin and PYY levels are associated with delayed gastric emptying and reduced gallbladder contractility(Reference Di Francesco, Zamboni and Dioli86). These high cholecystokinin and PYY levels facilitate long-lasting satiety and hunger suppression after meals and can lead to restriction of energy intake and malnutrition in the elderly(Reference Di Francesco, Zamboni and Dioli86).
Gastric inhibitory polypeptide
Gastric inhibitory polypeptide (GIP) is a forty-two-amino acid incretin peptide, which is released from endocrine K-cells in the duodenum and proximal jejunum within minutes after food ingestion(Reference Baggio and Drucker87). The main stimulus for GIP secretion is the presence of glucose and fat(Reference Creutzfeldt, Ebert and Willms88). GIP promotes energy storage by direct actions on adipose tissue. The peptides exert several anabolic adipocyte actions(Reference Creutzfeldt, Ebert and Willms88, Reference Salera, Giacomoni and Pironi89) as well as lipolytic effects. GIP-receptor-knock-out mice have lower adipocyte mass and display a resistance to diet-induced obesity(Reference Miyawaki, Yamada and Ban90). GIP on its own has no acute impact on food intake(Reference Baggio and Drucker87), but acts in concert with GLP-1 to control food intake and energy absorption. Similar to GLP-1, GIP increases glucose-dependent insulin secretion, β-cell proliferation and resistance to apoptosis(Reference Ding and Gromada91). GIP levels have been found to be elevated in obese individuals(Reference Baggio and Drucker87).
Gut hormones and appetite after bariatric surgery
Changes in appetite are evident within days of bariatric surgery(Reference le Roux, Welbourn and Werling10). Postprandial levels of gastrointestinal hormones that induce satiety, such as GLP-1 and PYY, are elevated after gastric bypass surgery(Reference Borg, le Roux and Ghatei92), but not after gastric banding(Reference le Roux, Aylwin and Batterham93). It has been shown that hunger is reduced and satiety is elevated if gastric bands are optimally inflated(Reference Dixon, Dixon and O'Brien13). These changes in appetite appear independent of any gut hormone alterations(Reference le Roux, Aylwin and Batterham93). Administration of octreotide, which would inhibit gut hormone responses, does not affect food intake after gastric banding(Reference le Roux, Aylwin and Batterham93). Thus, non-hormonal mechanisms have been suggested(Reference le Roux, Aylwin and Batterham93). In contrast, studies have demonstrated that postprandial PYY and GLP-1 levels start rising as early as 2 d after gastric bypass and can remain elevated for many months after surgery(Reference le Roux, Welbourn and Werling10, Reference Naslund, Gryback and Hellstrom11). In patients with only 20% weight loss after gastric-bypass operations the postprandial PYY and GLP-1 responses are attenuated compared with patients with 40% post-operative weight loss(Reference le Roux, Welbourn and Werling10). Moreover, inhibition of the satiety gastrointestinal hormone response with octreotide after gastric bypass increases appetite and food intake(Reference le Roux, Welbourn and Werling10). The proposed mechanism behind these findings is that bariatric surgery gives a secretory stimulus to the distal L-cells, resulting in an increased level of gastrointestinal hormones such as PYY and the enteroglucagon family of peptides(Reference le Roux, Aylwin and Batterham93). As a result, patients have long-term decreased appetite after gastric bypass. The combined effect of exogenous elevation of PYY and GLP-1 reduces food intake more than predicted by individual hormone infusions alone(Reference Neary, Small and Druce94). This combination of gastrointestinal hormone responses might, therefore, contribute to the successful weight loss and its maintenance after bariatric surgery.
On the other hand, changes in ghrelin levels after bariatric surgery are controversial. Ghrelin levels have been reported to be markedly suppressed after gastric bypass, while diet-induced weight loss is associated with increased levels of plasma ghrelin(Reference Cummings, Weigle and Frayo43). It was suggested that reduced ghrelin contributes to the weight loss after gastric bypass(Reference Cummings, Weigle and Frayo43). Other authors have published conflicting results(Reference Couce, Cottam and Esplen95–Reference Stenstrom, Zhao and Tommeras99). Thus, the role of ghrelin after gastric bypass remains unclear. Ghrelin secretion might in fact be modified by other gastrointestinal hormones, the levels of which change in response to the altered gastrointestinal anatomy. However, since obesity is associated with lower levels of ghrelin, it seems unlikely that reducing the level of ghrelin would, by itself, induce weight loss(Reference Rubino and Gagner100).
Long-term follow-up data on the changes in gastrointestinal hormones after bariatric surgery are still awaited. Surgery modulates a number of the gut hormones and probably allows them to act in concert in such a way as to affect appetite optimally. Understanding the contribution each hormone makes to appetite control within the setting of gastric-bypass surgery may be the stepping stone to future anti-obesity treatments.
Conclusions
Gastrointestinal hormones have attracted a remarkable amount of research interest in recent years because of their physiological effects on energy balance and appetite effects. Gastric bypass surgery is associated with elevated satiety and satiety-inducing gut hormones. Blocking these hormones reverses the satiety effects. Although surgery has been shown to be beneficial for the time being, it carries a risk for complications for patients. Bariatric surgery may thus be used as a model to understand physiological weight loss. This knowledge may help to guide future surgical and non-surgical weight-loss treatments.
Acknowledgements
The authors declare no conflict of interest. M. B. wrote the paper and carried out the literature review. C. W. le R. designed and carried out the research and analysed the data.


