


The increase in obesity and its associated diseases makes it more crucial than ever to better understand the mechanisms controlling food intake and energy metabolism. Feelings of hunger and satiety are key factors in controlling food intake. In normal individuals, there is a balance between the feeling of hunger before eating and the feeling of fullness that occurs after the assimilation of nutrients. This balance is deregulated in the context of obesity, in which the feeling of fullness is delayed or diminished(Reference Covasa1,Reference Little and Feinle-Bisset2) . Protein-enriched diets (PED) represent a particular nutritional situation exerting beneficial effects in glucose homeostasis in obese and diabetic patients. Dietary protein intake plays a beneficial role in glucose homeostasis, through the induction of satiety and weight loss in animals and human subjects(Reference Barkeling, Rössner and Björvell3–Reference Drummen, Tischmann and Gatta-Cherifi6). However, in mice models, manipulation of dietary protein levels (10–30 % by energy) at fixed fat contents (either 20 or 60 % by energy) has no effect on energy intake(Reference Hu, Wang and Yang7). Protein end products can be detected by several mechanisms during digestion, absorption and even during the inter-prandial period(Reference Fromentin, Darcel and Chaumontet8). Among them, anorexigenic gut peptides, such as cholecystokinin, glucagon-like peptide-1 and peptide YY, might be released after protein ingestion and target both directly and indirectly (mainly through the vagus nerve) the central nervous system to reduce food intake(Reference Fromentin, Darcel and Chaumontet8). In the long term, improvement in insulin sensitivity in diabetic patients is considered to be the result of weight loss(Reference Drummen, Tischmann and Gatta-Cherifi6). However, the parameters of glucose tolerance, including glycated Hb, are significantly improved in the short term in patients with type 2 diabetes, after increasing the proportion of dietary protein for a few weeks, independently of any effect on body weight(Reference Gannon and Nuttall9–Reference Gannon, Nuttall and Saeed11). The magnitude of the beneficial effects of PED depends on the amount and type of protein in the diet. Meta-analyses indicate that higher-protein diets containing between 1⋅2 and 1⋅6 g protein/kg/d and with meal-specific protein quantities of at least 25–30 g protein/meal provide a decrease in food intake, improvement in body weight management and/or cardiometabolic risk factors compared with lower-protein diets(Reference Leidy, Clifton and Astrup12).
The intestine and more particularly afferents to the brain of the extrinsic gastrointestinal nervous system (e.g. vagus nerve) are clearly involved in the control of feelings of hunger and satiety. We identified several years ago that enterocytes express the enzymatic machinery needed to produce glucose de novo and then demonstrated that the intestine is the third gluconeogenic organ, in addition to the liver and kidney. Results of the past 10 years have highlighted how intestinal gluconeogenesis (IGN) participates in whole-body metabolism. We here review how, by using nutritional (particularly PED) and genetic strategies targeting IGN, we identified numerous beneficial effects of this function on glucose and energy homeostasis. We also show how characterizing IGN could allow us to propose novel strategies for a beneficial use of PED on health.
Portal glucose and the regulation of food intake
The regulation of hunger could be artificially separated into two stages: satiation and satiety. It may be useful to recall here that satiation is defined as the cessation of the feeling of hunger that takes place during the absorption and digestion of the meal. In other words, satiation integrates the mechanisms that take place during the digestion of nutrients and help to reduce the feeling of hunger. The mechanisms underlying the shift from the sensation of hunger to satiation integrate the mechanisms that take place during the digestion of nutrients: gastric distension, changes in gut motility and secretion of gastrointestinal hormones such as ghrelin, cholecystokinin, peptide YY and glucagon-like peptide-1(Reference Janssen, Vanden Berghe and Verschueren13). In contrast, satiety is defined as the state of non-hunger that takes place following the digestion of the last meal. Thus, satiety moderates the feeling of hunger at the initiation of the next meal. Interestingly, dietary proteins mainly induce a feeling of satiety, not of satiation(Reference Rolls, Hetherington and Burley5). However, it is sometimes difficult to attribute a role of satiation or satiety for a given mechanism because a satiation mechanism can be prolonged and thus contribute to satiety.
Among the different signals originating from food, glucose has been identified as a key-signalling molecule able to suppress food intake(Reference Thorens and Larsen14) and drive the decision to eat(Reference Louis-Sylvestre and Le Magnen15). The digestion of a meal representative of current human nutrition (about 50 % of energy in the form of carbohydrates) leads to a high flow of glucose appearance in the portal vein. It might reach one to two times the equivalent of the total endogenous glucose production (EGP) of the body (excluding food). It has long been assumed that glucose could induce a satiating effect during the digestion of the meal. However, portal infusion of glucose at much lower flows (one-sixth to one-third of EGP) in previously unfed animals is sufficient to initiate both limitation of food intake and activation of the hypothalamic nuclei controlling the sensation of hunger in re-feeding period(Reference Delaere, Akaoka and De Vadder16,Reference Mithieux, Misery and Magnan17) . Conversely, various arguments have suggested that the release of glucose into the portal blood does not determine the end of a current meal, but instead reduces the size of the next meal(Reference Mithieux, Misery and Magnan17,Reference Baird, Grill and Kaplan18) . This has suggested that portal glucose may initiate satiety, rather than satiation.
The satiety effect of glucose infusion in the portal vein argues for the detection of glucose at a peripheral site. Indeed, after being proposed at the end of the 1960s, the presence of portal glucose sensors involved in the limitation of food intake has been abundantly documented, especially regarding the effect of intraportal infusions of glucose on food intake(Reference Langhans, Grossmann and Geary19–Reference Tordoff, Tluczek and Friedman22). The identification of nerve connections between the portal vein and the brain stem and the hypothalamic areas controlling food intake has further argued for a gut–brain signalling process induced by portal glucose detection(Reference Adachi, Shimizu and Oomura23,Reference Niijima24) . Finally, a large body of evidence allowed us to propose that the sodium-glucose co-transporter 3 could be responsible for portal glucose detection, rather than the GLUT2 or sweet taste receptors(Reference Delaere, Duchampt and Mounien25).
Intestinal gluconeogenesis and protein-enriched diet: induction of the portal glucose signal
Glucose release in the portal vein can originate from the meal but also from de novo synthesis (gluconeogenesis) from the intestine. Gluconeogenesis is a biological function relaying food to maintain blood glucose levels about 0⋅9–1 g/l. Glucose production into the bloodstream depends on the expression of the glucose-6-phosphatase (G6Pase), which catalyses the de-phosphorylation of glucose-6-phosphate into glucose (Fig. 1). The capacity of the liver and kidney to express this enzyme and to produce glucose was known since the end of the nineteenth century (for the liver) and the 1960s (for the kidney). However, we demonstrated at the end of the 1990s that the small intestine has the capacity to release glucose into the portal vein, during fasting and in the situation of insulin deficiency(Reference Croset, Rajas and Zitoun26–Reference Pillot, Soty and Gautier-Stein28). Before these data, one of the earlier demonstrations of intestinal G6Pase expression was performed from human jejunal mucosa(Reference Öckerman29). Moreover, during surgical operations in four patients, the conversion of fructose infused in the lumen into glucose released at the basolateral site was reported, which highlighted the functional character of the enzyme and of the gluconeogenesis pathway in human subjects(Reference Öckerman and Lundborg30).
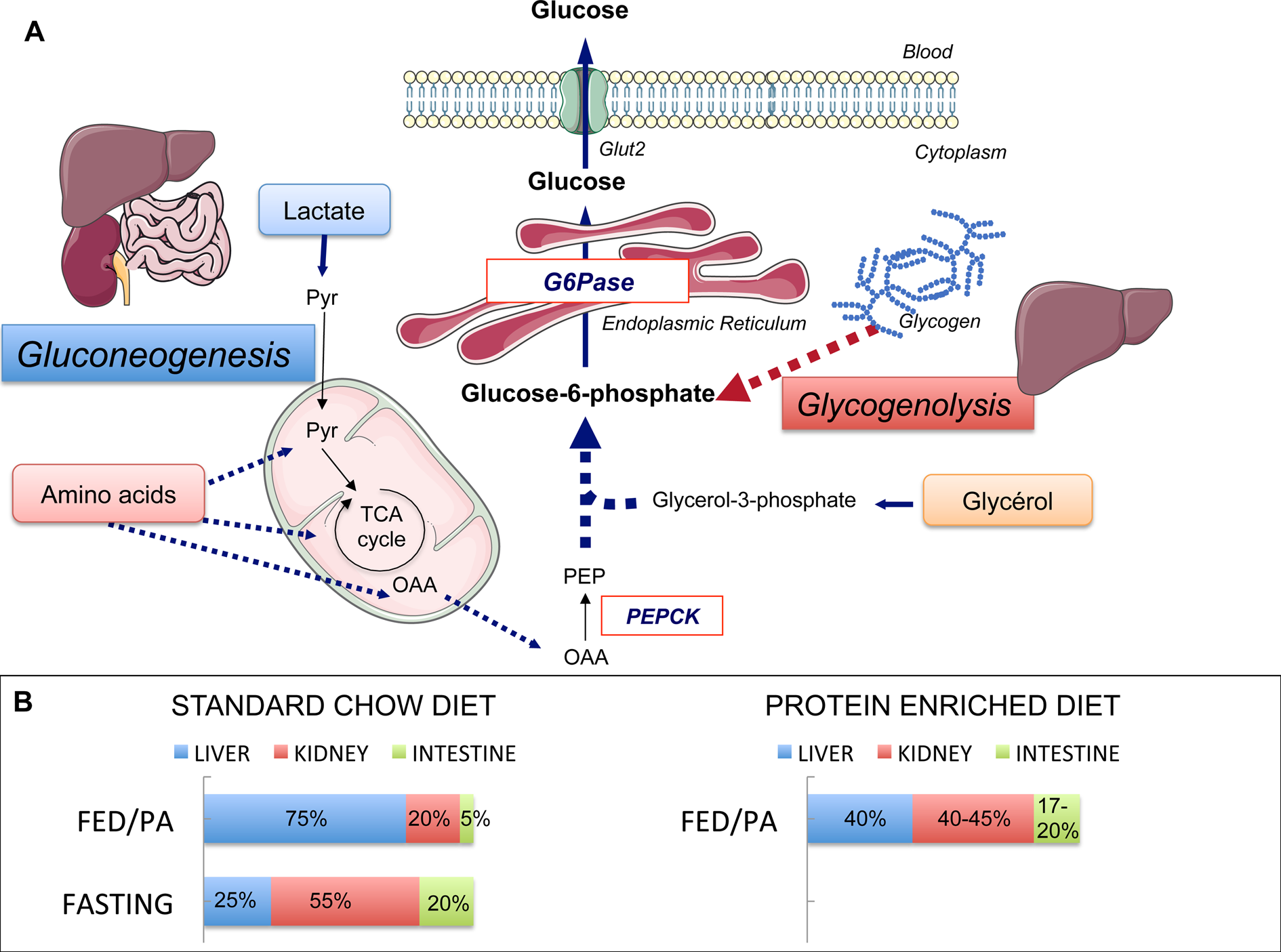
Fig. 1. Endogenous glucose production (EGP), main pathways and organs contribution. (a) Main pathways of EGP. Glycogenolysis in the liver and gluconeogenesis in the liver, kidney and intestine are the two pathways of EGP, both ending at the production of glucose-6-phosphate. The latter is produced from glycogen stores (in the liver only) or from lactate, amino acids and glycerol. Phosphoenolpyruvate carboxykinase (PEPCK) converts oxaloacetate (OAA) into phosphoenolpyruvate (PEP) and is considered as the rate-limiting enzyme of gluconeogenesis. Glucose-6-phosphatase (G6Pase) catalyses the hydrolysis of glucose-6-phosphate into glucose. G6Pase is the mandatory enzyme of EGP. The expression of its catalytic unit G6PC1 restricted to the liver, kidney and intestine confers on these organs their gluconeogenic capacities. Pyr, pyruvate; TCA, tricarboxylic. (b) Contribution of the gluconeogenic organs to EGP during standard chow or protein-enriched diet. The contribution of these three organs varies according to nutritional states. In the fed post-absorptive state (FED/PA) under a standard chow diet (left panel), the majority of endogenous glucose is produced by the liver. In the fasting state, the contribution of the kidney increases up to 55 % of total EGP at the expense of one of the liver. The intestine contributes to only 5–7 % of total EGP in the fed state on a standard chow diet, but makes a significant contribution to glucose production during fasting. A similar distribution of EGP among gluconeogenic organs is observed in the FED/PA under a protein-enriched diet (right panel). Servier Medical Art was used for illustrations.
Glucose production among gluconeogenic organs varies in function of nutritional situations. Under a standard starch-based diet, the liver provides the majority of EGP at the fed to fasted transition (i.e. the beginning of the post-absorptive period), while the kidney glucose production represents about 15–20 % of EGP in rats (Fig. 1). During fasting (24 h in rats), the kidney becomes the major contributor of EGP, providing about 50 % of EGP(Reference Pillot, Soty and Gautier-Stein28). Comparable estimations for both organs were obtained in human subjects in post-absorptive state(Reference Gerich, Meyer and Woerle31) and long-term fasting(Reference Owen, Felig and Morgan32). The intestine contributes to only 5–7 % of EGP at the fed to fasted transition but to about 20–25 % after 24 h fasting in the rat(Reference Croset, Rajas and Zitoun26,Reference Mithieux, Gautier-Stein and Rajas33) (Fig. 1). The contribution of IGN to glucose appearance in portal blood has now been firmly established during the an-hepatic phase of liver transplantation in human patients, where the kidney could account for about 70 % of EGP and the intestine for the remaining 30 %(Reference Battezzati, Caumo and Martino34), and after gastric bypass surgery in obese patients(Reference Hayes, Foo and Besic35). It could be estimated that IGN could account for at least 25 % of EGP in the latter patients(Reference Mithieux36).
The position of the intestine upstream of the portal nervous system has suggested that IGN might have the capacity to decrease hunger sensation by triggering the satiety effect of portal glucose. To provide this proof of concept, we used a nutritional strategy targeting IGN. Remarkably, a significant increase of IGN in the post-absorptive state (comparable to the contributions of the intestine to EGP in the fasted state; Fig. 1) takes place upon feeding PED(Reference Mithieux, Misery and Magnan17,Reference Mithieux37) . This increase is sufficient to counteract the high intestinal glucose utilisation, resulting in a portal glucose concentration equal to arterial blood glucose in the post-absorptive state (while it is substantially lower than arterial blood glucose after a high-carbohydrate diet). In this specific nutritional situation, the release of glucose into the portal vein by IGN is sufficient to activate the portal glucose sensor and to curb hunger and food intake(Reference Mithieux, Misery and Magnan17,Reference Mithieux37) . As expected, portal innervation is essential in this phenomenon, since local periportal treatment with capsaicin (a drug that inactivates both vagal- and spinal-sensitive nerves) abolishes the satiety effect induced by PED(Reference Mithieux, Misery and Magnan17). The causal link of IGN in the effect of dietary protein satiety has been confirmed by genetic manipulation of the gene encoding the catalytic unit of the G6Pase (G6pc) in mice. Thus, mice deleted for the G6pc gene specifically in the intestine are insensitive to the feeling of satiety induced by PED(Reference Penhoat, Mutel and Amigo-Correig38).
Mechanisms of induction of intestinal gluconeogenesis by proteins
PED promote IGN by inducing the expression of the intestinal gluconeogenic enzymes: G6Pase, phosphoenolpyruvate carboxykinase and glutaminase. Glutamine is indeed one of the major precursors of glucose synthesised by the intestine (Fig. 1; for review, see(Reference Mithieux27,Reference Mithieux37) ). Interestingly, denervation of the portal vein by capsaicin blunts these inductions by PED, suggesting that PED indirectly control IGN via a gut–brain axis.
The digestion of protein gives rise to the delivery of peptides into the portal blood after traversing the enterocyte mucosa(Reference Lee39). The infusion of peptides into the portal vein is sufficient to induce IGN but this process is blunted when portal-sensitive nerves are disrupted(Reference Duraffourd, De Vadder and Goncalves40). These results suggest that peptides can be detected at the level of the portal vein and may account for the control of food intake via IGN. Firstly, proteolytic fragments released from food proteins are known for a long time to exhibit μ-opioid activity in vitro (Reference Capasso, Amodeo and Balboni41–Reference Zioudrou, Streaty and Klee43). A large literature mentions the μ-opioid activity of oligopeptides of variable size, the minimum required structure being that of a dipeptide. Secondly, the modulation of μ-opioid receptors (MOR) controls food intake: agonists enhance food intake, whereas antagonists inhibit it (for review, see(Reference Yeomans and Gray44)). Third, MOR are present in the enteric nervous system(Reference Holzer45). This has led to the hypothesis that MOR may be involved in the detection of oligopeptides resulting from protein digestion and mediate the control of food intake by PED. In line with this hypothesis, MOR agonists (such as DAMGO) inhibit the expression of the regulatory genes of IGN and increase food intake when infused into the portal vein of conscious rats. In contrast, antagonists (such as naloxone) or peptides from different sources induce IGN and decrease food intake(Reference Duraffourd, De Vadder and Goncalves40). The causal effect of MOR in the control of hunger by proteins related to IGN has been confirmed by knockout experiments. MOR-knockout mice do not induce IGN in response to oligopeptides and are insensitive to PED. Moreover, mice with intestinal G6pc1 deletion do not decrease food intake in response to portal infusions of MOR antagonists or peptides(Reference Duraffourd, De Vadder and Goncalves40).
To sum-up, PED induces two consecutive gut–brain signals initiated at the level of the portal vein (Fig. 2). The first detection of peptides by MOR starts at the post-prandial period and leads to the induction of IGN by a gene induction process. This is progressive and takes place over the entire postprandial period. Following the induction of IGN, the portal glucose signal can initiate satiety during the post-absorptive period (Fig. 2). This may continue after the postprandial period, since it depends on robust induction at the enzyme level(Reference Duraffourd, De Vadder and Goncalves40). Moreover, glutamine and glutamate, major substrates of IGN, are present at high concentration in the blood in all nutritional situations. Therefore, deciphering the role of IGN in the effect of protein diets has provided a mechanistic explanation for their effect of satiety.
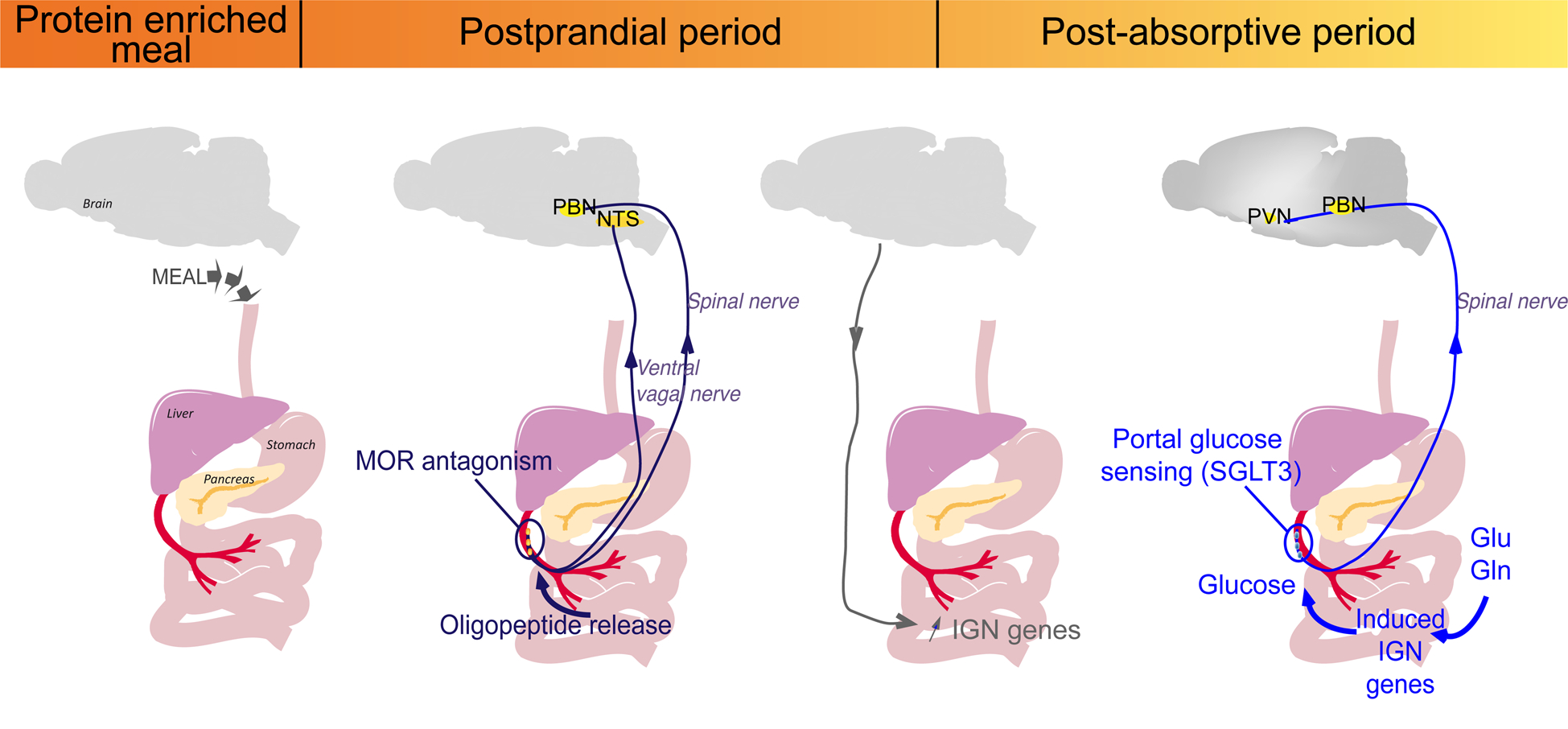
Fig. 2. Sequential activation of intestinal gluconeogenesis (IGN) after a protein-enriched meal. The digestion of protein-enriched meal leads to the release of peptides in the portal vein. The antagonist effect of peptides on μ-opioid receptors (MOR) during the post-prandial period activates a gut–brain signal transmitted by the vagal and spinal nerves. Then, a brain–gut neural signal induces the regulatory genes of IGN. This is progressive and takes place over the entire postprandial period. During the post-absorptive period, glucose can be produced from gluconeogenic substrates (glutamine (Gln) or Glutamate (Glu) from protein digestion or from the blood), released in the portal vein and detected by the sodium-glucose co-transporter 3 (SGLT3) receptor. This portal glucose signal induces neuronal activity in the parabrachial nucleus (PBN) through the spinal nerves and in the hypothalamus (PVN, paraventricular nucleus). This may continue after the postprandial period, since it depends on robust induction at the enzyme level, and the permanent availability of IGN substrates, such as Gln or Glu(Reference Duraffourd, De Vadder and Goncalves40). NTS, nucleus of the solitary tract.
Brain targets of portal glucose signal and protein diets
Hunger is determined at the level of the brain, which integrates circulating and nervous signal from the periphery. The hypothalamus and brain stem are the regions historically most studied for the control of food intake and energy metabolism.
The regions of the brain stem that receive vagal afferents (i.e. the dorsal vagal complex) and those that receive spinal afferents (i.e. the parabrachial nucleus) are both activated by PED (as revealed by C-FOS labelling in rats), suggesting that they are both involved in the reflex arc initiated by IGN induction(Reference Fromentin, Darcel and Chaumontet8,Reference Delaere, Akaoka and De Vadder16,Reference Mithieux, Misery and Magnan17) . However, the surgical disruption of either the vagal or the spinal afferents to the brain suggests that portal glucose signalling is conveyed to the central nervous system by the spinal route and not by the vagal route(Reference Delaere, Akaoka and De Vadder16,Reference Delaere, Duchampt and Mounien25) . Infusion of portal glucose activates the expression of C-FOS only in the parabrachial nucleus(Reference Delaere, Akaoka and De Vadder16), and not in the dorsal vagal complex, which confirms the surgical data.
In addition to the parabrachial nucleus, PED and portal glucose infusion also activate the expression of C-FOS in the area of the hypothalamus involved in the control of food intake, such as the arcuate nucleus(Reference Delaere, Akaoka and De Vadder16,Reference Faipoux, Tomé and Gougis46) . This region contains the first-order neurons of the melanocortinergic system, which is the main regulator of food intake and energy homeostasis in the hypothalamus. Neurons present in the arcuate nucleus have specific access to nutrients and hormones of the blood flow via the fenestrated capillaries of the median eminence. More precisely, neurons co-expressing proopiomelanocortin and cocaine-amphetamine-related transcript rapidly respond to nutritional information by inducing anorexigenic signals. Proopiomelanocortin is cleaved into melanocyte-stimulating hormone, which exerts anorectic stimuli by binding to melanocortin receptors (MC3 and MC4R) on second-order neurons of the paraventricular nucleus. Conversely, neurons co-expressing neuropeptide Y and agouti-related protein induce orexigenic signals. Neuropeptide Y/agouti-related protein neurons have an opposite effect to proopiomelanocortin/cocaine-amphetamine-related neurons through the antagonism of agouti-related protein on MC3R and MC4R. PED and portal glucose infusion increase the proportion of activated neurons in the arcuate nucleus, suggesting the involvement of the melanocortinergic system in their effect on food intake(Reference Delaere, Akaoka and De Vadder16,Reference Faipoux, Tomé and Gougis46) .
Finally, both PED and portal glucose signal also induce C-FOS activation in olfactory and other limbic and cortical areas, including those functionally implicated in reward(Reference Delaere, Akaoka and De Vadder16,Reference Darcel, Fromentin and Raybould47) . In addition to satiety and metabolic effects primarily identified, IGN primed by PED may thus influence behavioural adaptation via a network including the hypothalamus and the sensory and corticolimbic systems(Reference Delaere, Akaoka and De Vadder16). Consistently, high-protein diet was shown to modulate the reactivity of corticolimbic brain area (hippocampus, orbitofrontal cortex and striatum) in response to food cues in women(Reference Leidy, Ortinau and Douglas48,Reference Griffioen-Roose, Smeets and van den Heuvel49) .
Beneficial effects of protein-enriched diet and intestinal gluconeogenesis on whole-body metabolism
Increasing the proportion of dietary proteins for a few weeks markedly improves glucose tolerance in the short term in type 2 diabetic patients without any effect on body weight(Reference Gannon and Nuttall9,Reference Gannon, Nuttall and Saeed11) .
Precisely, increasing the amount of protein in the diet from 15 to 30 % (with a parallel decrease of carbohydrates from 55 to 40 %) during 5 weeks only decreased the fasting glucose response by 38 % and the percentage of total glycohaemoglobin from 8⋅1 to 7⋅3. These clinical studies suggest a beneficial effect of PED on glucose homeostasis independently of their effect on food intake and body weight management(Reference Gannon and Nuttall10,Reference Layman, Clifton and Gannon50) .
Besides increasing IGN, PED leads to a redistribution of EGP among gluconeogenic organs (Fig. 1)(Reference Pillot, Soty and Gautier-Stein28). PED induces the expression of the key gluconeogenic genes in the kidney leading to an increase in renal gluconeogenesis up to about 45 % of total EGP(Reference Pillot, Soty and Gautier-Stein28). Both renal gluconeogenesis and IGN are induced without a global increase in EGP, suggesting a reduction in liver glucose production. It must be recalled here the opposite effects of liver and intestinal gluconeogeneses on glucose homeostasis. On the contrary to the benefits conferred by IGN, increased hepatic gluconeogenesis (HGN) is deleterious for glucose control. HGN is increased in type 2 diabetic patients(Reference Granner and O'Brien51,Reference Magnusson, Rothman and Katz52) and increased HGN in genetically manipulated rats is sufficient to initiate insulin resistance(Reference Trinh, O'Doherty and Anderson53). Consistently, the suppression of HGN by targeted deletion of G6Pase in the liver confers strong protection against the development of diabetes induced by a high-fat/high-sucrose diet(Reference Abdul-Wahed, Gautier-Stein and Casteras54). Therefore, HGN and IGN exert opposite effects on glucose control: increased HGN promotes metabolic anomalies, whereas IGN promotes metabolic benefits via portal glucose signalling. Given the deleterious role of HGP in glucose homeostasis, the reduction in liver glucose production by PED may impact insulin sensitivity and glucose homeostasis. Indeed, PED markedly ameliorates the suppression of EGP by insulin whereas having no effect on peripheral glucose uptake(Reference Pillot, Soty and Gautier-Stein28). More precisely, insulin suppression of EGP in PED-fed rats concerns mainly the glycogenolysis pathway. In keeping with this proposal, hepatic glycogen stores are higher in PED-fed rats upon insulin stimulation. In agreement with a causal role of IGN in the beneficial effect of PED on insulin sensitivity, mice lacking IGN develop a pre-diabetic state while fed a conventional diet, including elevated fasting glucose and insulin concentrations, glucose intolerance and insulin resistance, and defective insulin secretion in response to glucose(Reference Soty, Penhoat and Amigo-Correig55). Moreover, they are prone to diabetes since they become diabetic much more rapidly than their control counterparts under a high-fat/high-sucrose diet(Reference Soty, Penhoat and Amigo-Correig55). As for the regulation of food intake by PED, our results provide a mechanistic explanation for the rapid and spectacular improvement of glucose homeostasis observed in type 2 diabetic patients, upon increasing the proportion of protein in their diet(Reference Gannon and Nuttall9,Reference Gannon, Nuttall and Saeed11) .
Future directions
By characterizing the link between PED and IGN, we identified the mechanism underlying the satiety effect of proteins. Then, studies on mice model of GM IGN have allowed us to identify the different functions controlled by the portal glucose signal(Reference Soty, Gautier-Stein and Rajas56). However, several steps must be completed before the validation of a therapeutic strategy in human health.
In terms of basic research, determining whether proteins of different origins have the same effect on IGN could help guide choices in nutritional recommendations. Proteins are metabolised by the microbiota to produce SCFA (which are associated with beneficial effects on health) but also other components that may promote intestinal inflammation and colorectal cancer(Reference Diether and Willing57). The amount of these beneficial/deleterious bioactive end-products may vary in function of the nature of the protein in the diet(Reference Diether and Willing57,Reference Blachier, Beaumont and Portune58) . The simultaneous analyses of microbiota and metabolites from feces of PED-fed mice should help us to identify the beneficial compounds from protein digestion, which exhibit increasing specificity and efficiency to induce IGN. These results should help in establishing nutritional recommendations that improve metabolic outcomes and have no impact on the gut environment.
The gold standard method to measure IGN is based on glucose radioactive tracers and the estimation of their specific activity in the artery and vein surrounding the intestine, i.e. the systemic and portal blood, respectively, in our experiments. In human subjects, access to portal blood is possible in specific situations, mainly during surgeries such as gastric by-pass or liver transplantation, which has permitted to confirm the gluconeogenic capacity of the human intestine(Reference Battezzati, Caumo and Martino34–Reference Mithieux36). However, blood sampling from the portal vein of healthy individual presents medical dangers. Consequently, the currently available techniques do not allow us to monitor IGN in patients and to validate and further document the beneficial effects of this function on human health. Then, the specific mouse models we developed in the laboratory should be used to solve this issue. Untargeted MS methods applied to mouse with a genetic deletion or induction of IGN should help us to identify circulating biomarkers in relation with IGN. Such markers could then be used in translational studies to assess the role of IGN in human metabolism. The same markers could help us to assess the capacity of diet, nutrients or even drugs to induce IGN and mediate its beneficial effect on human health.
Conclusions
The use of PED in the improvement of the metabolic status of obese and diabetic patients is still a matter of debate(Reference Drummen, Tischmann and Gatta-Cherifi6,Reference Portune, Beaumont and Davila59,Reference Ullah, Rauf and Nabi60) and the mechanisms of these beneficial effects are not completely identified. Using rodent models, we identify IGN and a gut–brain signal triggered by protein digestion as the mechanism underlying the satiety effect of proteins. Using the same models, we highlighted that IGN is the mechanistic link of the beneficial effect of protein-enriched diet on plasma glucose control. While the gluconeogenic capacity of the intestine has been demonstrated in human subjects, the lack of current techniques allowing a longitudinal measurement of IGN in patients prevents the development of translational studies. Further studies on the links between PED and IGN and new IGN measurement techniques should pave the way for the identification of new nutritional approaches to prevent the serious metabolic consequences of both obesity and diabetes.
Acknowledgement
The authors thank all laboratory members who have contributed to these works. The authors thank the Centre National de la Recherche Scientifique (F. R. and G. M.) and the Institut National de la Recherche Agronomique (A. G. -S.) for funding their positions. Additionally, the Institut National de la Santé et de la Recherche Médicale and the Université Lyon 1 provided laboratory funding and housing.
Financial Support
This work received financial support from the Agence Nationale de la Recherche (ANR11-BSV1-016-01), Fondation pour la Recherche Medicale (DRM20101220448 and Equipe FRM DEB20160334898), Institut Delessert, Institut Bonduelle and Société Francophone du Diabète.
Conflict of Interest
None.
Authorship
The authors had joint responsibility for all aspects of preparation of this paper.


