
107 results
2 - Continuum Mechanics
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 60-109
-
- Chapter
- Export citation
10 - Viscoelasticity
- from Part I - Physical Tools
-
- Book:
- The Physics of Bacteria
- Published online:
- 12 December 2024
- Print publication:
- 19 December 2024, pp 89-94
-
- Chapter
- Export citation
Atomic molecular dynamics simulation advances of de novo-designed proteins
-
- Journal:
- Quarterly Reviews of Biophysics / Volume 57 / 2024
- Published online by Cambridge University Press:
- 05 December 2024, e14
-
- Article
-
- You have access
- HTML
- Export citation
Slip due to kink propagation at the liquid–solid interface
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A19
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Effect of layer bending on montmorillonite hydration and structure from molecular simulation
-
- Journal:
- Clays and Clay Minerals / Volume 72 / 2024
- Published online by Cambridge University Press:
- 20 November 2024, e24
-
- Article
-
- You have access
- HTML
- Export citation
Simulations of Interlayer Methanol in Ca- and Na-Saturated Montmorillonites Using Molecular Dynamics
-
- Journal:
- Clays and Clay Minerals / Volume 49 / Issue 3 / June 2001
- Published online by Cambridge University Press:
- 28 February 2024, pp. 255-262
-
- Article
-
- You have access
- Export citation
Project 4: - Planetary System
-
- Book:
- A First Guide to Computational Modelling in Physics
- Published online:
- 01 February 2024
- Print publication:
- 08 February 2024, pp 26-33
-
- Chapter
- Export citation
Intercalation of Ethylene Glycol in Smectites: Several Molecular Simulation Models Verified by X-Ray Diffraction Data
-
- Journal:
- Clays and Clay Minerals / Volume 64 / Issue 4 / August 2016
- Published online by Cambridge University Press:
- 01 January 2024, pp. 488-502
-
- Article
-
- You have access
- Export citation
Molecular Dynamics Simulations of Anion Exclusion in Clay Interlayer Nanopores
-
- Journal:
- Clays and Clay Minerals / Volume 64 / Issue 4 / August 2016
- Published online by Cambridge University Press:
- 01 January 2024, pp. 374-388
-
- Article
-
- You have access
- Export citation
Monte Carlo and Molecular Dynamics Simulation of Uranyl Adsorption on Montmorillonite Clay
-
- Journal:
- Clays and Clay Minerals / Volume 51 / Issue 4 / August 2003
- Published online by Cambridge University Press:
- 01 January 2024, pp. 372-381
-
- Article
-
- You have access
- Export citation
-
We performed Monte Carlo and molecular dynamics simulations to investigate the interlayer structure of a uranyl-substituted smectite clay. Our clay model is a dioctahedral montmorillonite with negative charge sites in the octahedral sheet only. We simulated a wide range of interlayer water content (0 mg H2O/g clay — 260 mg H2O/g clay), but we were particularly interested in the two-layer hydrate that has been the focus of recent X-ray absorption experiments. Our simulation results for the two-layer hydrate of uranyl-montmorillonite yield a water content of 160 mg H2O/g clay and a layer spacing of 14.66 Å. Except at extremely low water content, uranyl cations are oriented nearly parallel to the surface normal in an outer-sphere complex. The first coordination shell consists of five water molecules with an average U-O distance of 2.45 Å, in good agreement with experimental data. At low water content, the cations can assume a perpendicular orientation to include surface oxygen atoms in the first coordination shell. Our molecular dynamics results show that
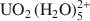 complexes translate within the clay pore through a jump diffusion process, and that first-shell water molecules are exchangeable and interchangeable.
complexes translate within the clay pore through a jump diffusion process, and that first-shell water molecules are exchangeable and interchangeable.
Ion Adsorption at Clay-Mineral Surfaces: The Hofmeister Series for Hydrated Smectite Minerals
-
- Journal:
- Clays and Clay Minerals / Volume 64 / Issue 4 / August 2016
- Published online by Cambridge University Press:
- 01 January 2024, pp. 472-487
-
- Article
-
- You have access
- Export citation
Molecular Dynamics Simulation of Alkylammonium-Intercalated Vermiculites
-
- Journal:
- Clays and Clay Minerals / Volume 65 / Issue 6 / December 2017
- Published online by Cambridge University Press:
- 01 January 2024, pp. 378-386
-
- Article
-
- You have access
- Export citation
Titratable residues that drive RND efflux: Insights from molecular simulations
-
- Journal:
- QRB Discovery / Volume 5 / 2024
- Published online by Cambridge University Press:
- 01 April 2024, e5
- Print publication:
- 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Diffusion of Na and Cs in montmorillonite
-
- Journal:
- Clays and Clay Minerals / Volume 56 / Issue 2 / April 2008
- Published online by Cambridge University Press:
- 01 January 2024, pp. 190-206
-
- Article
-
- You have access
- Export citation
Montmorillonite as an Anti-Tuberculosis Rifampicin Drug Carrier: DFT and Experimental Study
-
- Journal:
- Clays and Clay Minerals / Volume 71 / Issue 2 / April 2023
- Published online by Cambridge University Press:
- 01 January 2024, pp. 229-241
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Comparative Modeling of Ions and Solvent Properties in Ca-Na Montmorillonite by Atomistic Simulations and Fluid Density Functional Theory
-
- Journal:
- Clays and Clay Minerals / Volume 68 / Issue 2 / April 2020
- Published online by Cambridge University Press:
- 01 January 2024, pp. 100-114
-
- Article
-
- You have access
- HTML
- Export citation
Structure and Dynamics of Water—Smectite Interfaces: Hydrogen Bonding and the Origin of the Sharp O-Dw/O—Hw Infrared Band From Molecular Simulations
-
- Journal:
- Clays and Clay Minerals / Volume 64 / Issue 4 / August 2016
- Published online by Cambridge University Press:
- 01 January 2024, pp. 452-471
-
- Article
-
- You have access
- Export citation
Modeling the response of pyrophyllite interlayer to applied stress using steered molecular dynamics
-
- Journal:
- Clays and Clay Minerals / Volume 53 / Issue 2 / April 2005
- Published online by Cambridge University Press:
- 01 January 2024, pp. 171-178
-
- Article
-
- You have access
- Export citation
Molecular Simulation of Cesium Adsorption at the Basal Surface of Phyllosilicate Minerals
-
- Journal:
- Clays and Clay Minerals / Volume 64 / Issue 4 / August 2016
- Published online by Cambridge University Press:
- 01 January 2024, pp. 389-400
-
- Article
-
- You have access
- Export citation
A constitutive relation generalizing the Navier–Stokes theory to high-rate regimes
-
- Journal:
- Journal of Fluid Mechanics / Volume 974 / 10 November 2023
- Published online by Cambridge University Press:
- 09 November 2023, A30
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
