
74 results
Association of dietary manganese intake and the IL1R1 rs3917225 polymorphism with thyroid cancer risk: a prospective cohort study in Korea
-
- Journal:
- British Journal of Nutrition / Volume 132 / Issue 12 / 28 December 2024
- Published online by Cambridge University Press:
- 13 November 2024, pp. 1584-1592
- Print publication:
- 28 December 2024
-
- Article
-
- You have access
- HTML
- Export citation
Role of Manganese in the Oxidation of Arsenite by Freshwater Lake Sediments
-
- Journal:
- Clays and Clay Minerals / Volume 29 / Issue 3 / June 1981
- Published online by Cambridge University Press:
- 01 July 2024, pp. 219-225
-
- Article
-
- You have access
- Export citation
Manganese Minerals in Clays: A Review
-
- Journal:
- Clays and Clay Minerals / Volume 28 / Issue 5 / October 1980
- Published online by Cambridge University Press:
- 01 July 2024, pp. 346-354
-
- Article
-
- You have access
- Export citation
Influence of Mn2+ and pH on the Formation of Iron Oxides from Ferrous Chloride and Ferrous Sulfate Solutions
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 5 / October 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 451-458
-
- Article
-
- You have access
- Export citation
Refinement of Mn-Substituted Muscovite and Phlogopite
-
- Journal:
- Clays and Clay Minerals / Volume 34 / Issue 1 / February 1986
- Published online by Cambridge University Press:
- 02 April 2024, pp. 7-16
-
- Article
-
- You have access
- Export citation
Influence of Manganese Oxide Minerals on the Formation of Iron Oxides
-
- Journal:
- Clays and Clay Minerals / Volume 36 / Issue 5 / October 1988
- Published online by Cambridge University Press:
- 02 April 2024, pp. 467-475
-
- Article
-
- You have access
- Export citation
High-Resolution Transmission Electron Microscopy Study of Mn-Oxyhydroxide Transformations and Accompanying Phases in a Lateritic Profile of Moanda, Gabon
-
- Journal:
- Clays and Clay Minerals / Volume 39 / Issue 3 / June 1991
- Published online by Cambridge University Press:
- 02 April 2024, pp. 254-263
-
- Article
-
- You have access
- Export citation
Oxidation of 1,2- and 1,4-Dihydroxybenzene by Birnessite in Acidic Aqueous Suspension
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 5 / October 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 479-486
-
- Article
-
- You have access
- Export citation
Scanning Electron Microscopic and X-ray Powder Diffraction Study of Manganiferous Bauxite, Kincsesbánya, Hungary
-
- Journal:
- Clays and Clay Minerals / Volume 33 / Issue 6 / December 1985
- Published online by Cambridge University Press:
- 02 April 2024, pp. 532-538
-
- Article
-
- You have access
- Export citation
Characterization of Goethite and Hematite in a Tunisian Soil Profile By Mössbauer Spectroscopy
-
- Journal:
- Clays and Clay Minerals / Volume 34 / Issue 3 / June 1986
- Published online by Cambridge University Press:
- 02 April 2024, pp. 275-280
-
- Article
-
- You have access
- Export citation
Electron Transfer Processes Between Hydroquinone and Hausmannite (Mn3O4)
-
- Journal:
- Clays and Clay Minerals / Volume 36 / Issue 4 / August 1988
- Published online by Cambridge University Press:
- 02 April 2024, pp. 297-302
-
- Article
-
- You have access
- Export citation
Transformation of Akaganéite Into Goethite and Hematite in the Presence of Mn
-
- Journal:
- Clays and Clay Minerals / Volume 39 / Issue 2 / April 1991
- Published online by Cambridge University Press:
- 02 April 2024, pp. 144-150
-
- Article
-
- You have access
- Export citation
Effect of Cysteine and Manganese on the Crystallization of Noncrystalline Iron(III) Hydroxide at pH 8
-
- Journal:
- Clays and Clay Minerals / Volume 38 / Issue 1 / February 1990
- Published online by Cambridge University Press:
- 02 April 2024, pp. 21-28
-
- Article
-
- You have access
- Export citation
Mn-Substituted Goethite and Fe-Substituted Groutite Synthesized At Acid pH1
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 2 / April 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 151-156
-
- Article
-
- You have access
- Export citation
Weathering Sequence and Alteration Products in the Genesis of the Graskop Manganese Residua, Republic of South Africa
-
- Journal:
- Clays and Clay Minerals / Volume 36 / Issue 5 / October 1988
- Published online by Cambridge University Press:
- 02 April 2024, pp. 448-454
-
- Article
-
- You have access
- Export citation
Effect of Manganese on the Transformation of Ferrihydrite into Goethite and Jacobsite in Alkaline Media
-
- Journal:
- Clays and Clay Minerals / Volume 35 / Issue 1 / February 1987
- Published online by Cambridge University Press:
- 02 April 2024, pp. 11-20
-
- Article
-
- You have access
- Export citation
Transformation of Hausmannite into Birnessite in Alkaline Media
-
- Journal:
- Clays and Clay Minerals / Volume 36 / Issue 3 / June 1988
- Published online by Cambridge University Press:
- 02 April 2024, pp. 249-257
-
- Article
-
- You have access
- Export citation
Oxidation of Dihydroxybenzenes in Aerated Aqueous Suspensions of Birnessite
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 4 / August 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 341-347
-
- Article
-
- You have access
- Export citation
Melt synthesis and characterization of synthetic Mn-rich tainiolite
-
- Journal:
- Clays and Clay Minerals / Volume 57 / Issue 2 / April 2009
- Published online by Cambridge University Press:
- 01 January 2024, pp. 271-277
-
- Article
-
- You have access
- Export citation
-
Large transition-metal contents add desirable physical properties, such as redox reactivity, magnetism, and electric or ionic conductivity to micas and make them interesting for a variety of materials-science applications. A Mn- and F-rich tainiolite mica,
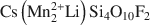 , was synthesized by a high-temperature melt-synthesis technique. Subsequent annealing for 10 days led to a single-phase and coarsegrained material. Single-crystal X-ray diffraction studies were performed and characteristic geometric parameters were compared to the analogous ferrous compound, synthetic Fe-rich tainiolite,
, was synthesized by a high-temperature melt-synthesis technique. Subsequent annealing for 10 days led to a single-phase and coarsegrained material. Single-crystal X-ray diffraction studies were performed and characteristic geometric parameters were compared to the analogous ferrous compound, synthetic Fe-rich tainiolite, 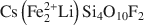 . Both tainiolite structures are outside the compositional stability limits for the 2:1 layer structure, and incorporating the relatively large cation Mn2+ requires significant structural adjustments in both the octahedral and tetrahedral sheets. As expected, increasing the ionic radius of the octahedral cation from 0.78 Å (VIFe2+) to 0.83 Å (VIMn2+) reduces the octahedral flattening angle from <Ψ> = 57.05° to <Ψ> = 56.4°, the smallest value ever observed for a tetrasilicic mica. However, even this small <Ψ> value is insufficient to match the lateral sizes of the tetrahedral and octahedral sheets and, in addition, unusual structural adjustments in the tetrahedral sheet are required. The average tetrahedral bond length <T-O> is much greater (1.643 Å) than the average value observed for tetrasilicic micas (1.607 Å,) and a significant difference between the <T-O>apical (1.605 Å) and the <T-O>basal bond lengths (1.656 Å) and an enlarged basal flattening angle (τbas = 106.29°) are noted. These parameters indicate: (1) that the 2:1 layer might be more flexible than previously thought, to allow matching of the lateral dimensions of the tetrahedral and octahedral sheets; and (2) that many other compositions that appear interesting from a materials-science point of view might be accessible.
. Both tainiolite structures are outside the compositional stability limits for the 2:1 layer structure, and incorporating the relatively large cation Mn2+ requires significant structural adjustments in both the octahedral and tetrahedral sheets. As expected, increasing the ionic radius of the octahedral cation from 0.78 Å (VIFe2+) to 0.83 Å (VIMn2+) reduces the octahedral flattening angle from <Ψ> = 57.05° to <Ψ> = 56.4°, the smallest value ever observed for a tetrasilicic mica. However, even this small <Ψ> value is insufficient to match the lateral sizes of the tetrahedral and octahedral sheets and, in addition, unusual structural adjustments in the tetrahedral sheet are required. The average tetrahedral bond length <T-O> is much greater (1.643 Å) than the average value observed for tetrasilicic micas (1.607 Å,) and a significant difference between the <T-O>apical (1.605 Å) and the <T-O>basal bond lengths (1.656 Å) and an enlarged basal flattening angle (τbas = 106.29°) are noted. These parameters indicate: (1) that the 2:1 layer might be more flexible than previously thought, to allow matching of the lateral dimensions of the tetrahedral and octahedral sheets; and (2) that many other compositions that appear interesting from a materials-science point of view might be accessible.
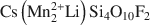 , was synthesized by a high-temperature melt-synthesis technique. Subsequent annealing for 10 days led to a single-phase and coarsegrained material. Single-crystal X-ray diffraction studies were performed and characteristic geometric parameters were compared to the analogous ferrous compound, synthetic Fe-rich tainiolite,
, was synthesized by a high-temperature melt-synthesis technique. Subsequent annealing for 10 days led to a single-phase and coarsegrained material. Single-crystal X-ray diffraction studies were performed and characteristic geometric parameters were compared to the analogous ferrous compound, synthetic Fe-rich tainiolite, 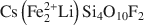 . Both tainiolite structures are outside the compositional stability limits for the 2:1 layer structure, and incorporating the relatively large cation Mn
. Both tainiolite structures are outside the compositional stability limits for the 2:1 layer structure, and incorporating the relatively large cation MnAn Electron Paramagnetic Resonance Spectroscopy Investigation of the Retention Mechanisms of Mn and Cu in the Nanopore Channels of Three Zeolite Minerals
-
- Journal:
- Clays and Clay Minerals / Volume 60 / Issue 6 / December 2012
- Published online by Cambridge University Press:
- 01 January 2024, pp. 588-598
-
- Article
-
- You have access
- Export citation
