Constant pressure molecular dynamics (MD) simulations have been used to simulate amorphous and crystalline forms of BaTiO3 and TiO2. Simulation results for pure RaTiO3 and TiO2 glasses show fourfold titanium coordination with a Ti–O bond distance of 1.8 Å, consistent with experimental evidence for the structure of titania doped glasses for all optical switching and ultra low expansivity (ULE) TiO2–SiO2 glasses. Radial distribution function data for crystalline, liquid, and amorphous forms of BaTiO3 were also obtained. The displacement polarization and its contribution to susceptibilities have been calculated by application of an electric field to the simulation cell. The calculated ionic dielectric constants (Ki) for simulated NaCI (crystal), TiO2 glass, and TiO2 (crystal) were 3.34, 5.96, and 19.4 as compared to the experimental values of ≍3.34, 3–10, and 78, respectively. The calculated cubic nonlinear susceptibility (ξ3) values for NaCI (crystal), TiO2 (glass), and TiO2 (crystal) were, respectively, 0.194, 2.175, and 4.68 (x 10−18m2V−2). The increase in ξ3 values is consistent with experimentally observed trends of the linear refractive indices. Improved agreement between experimental and calculated values of susceptibilities and dielectric constant was obtained for materials with higher ionicity.
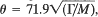 , where M is the atomic weight. This relation is derived theoretically in the Debye isotropic approximation by assuming that the interatomic potential is central. No restrictions are imposed on the range of the potential. The relation is obeyed very well by the observed values of θ and Γ in the case of many solids.
, where M is the atomic weight. This relation is derived theoretically in the Debye isotropic approximation by assuming that the interatomic potential is central. No restrictions are imposed on the range of the potential. The relation is obeyed very well by the observed values of θ and Γ in the case of many solids.